
Grantee Research Project Results
2016 Progress Report: Long Term Metabolic Consequences of Exposures to Multipollutant Atmospheres in the Great Lakes Region
EPA Grant Number: R834797C003Subproject: this is subproject number 003 , established and managed by the Center Director under grant R834797
(EPA does not fund or establish subprojects; EPA awards and manages the overall grant for this center).
Center: Center for Research on Early Childhood Exposure and Development in Puerto Rico
Center Director: Alshawabkeh, Akram
Title: Long Term Metabolic Consequences of Exposures to Multipollutant Atmospheres in the Great Lakes Region
Investigators: Rajagopalan, Sanjay , Sun, Qinghua
Institution: The Ohio State University
EPA Project Officer: Chung, Serena
Project Period: December 1, 2010 through November 30, 2015 (Extended to December 31, 2016)
Project Period Covered by this Report: December 1, 2015 through November 30,2016
RFA: Clean Air Research Centers (2009) RFA Text | Recipients Lists
Research Category: Human Health , Air
Objective:
We have recently demonstrated that short-term exposure to inhaled concentrated airborne particulate (CAP) matter <2.5µm (PM2.5) results in components of cardiometabolic syndrome (CMS) including development of hypertension and insulin resistance. In this project, we hypothesize that chronic inhalation of CAP in conjunction with gaseous components such as ozone from distinct multipollutant atmospheres synergistically interacts with diet and genetic susceptibility to influence development of CMS. Project 3 is an integral component of the overarching theme of this center that primary air pollutants, fine PM (PM2.5) and ozone (O3), cause cardiometabolic health effects that are dependent on the local atmospheric multipollutant milieu, predisposing factors, and the interactive toxicity of multipollutant coexposure. The experiments proposed are natural extensions of human research outlined in Project 1 and acute experiments in Project 2 and will focus on conducting chronic inhalation toxicology studies in diet fed and genetic models of obesity/diabetes. In Aim 1, simultaneous chronic exposure to multipollutant CAP from two locations in Columbus, OH representing near-roadside/traffic or remotely transported/aged emissions will be examined in combination with high fat chow (HFC). The impact of CAP on glucose/insulin homeostasis, adipokines, insulin signaling, adipose and pulmonary inflammation and an analysis of dose dependence and CAP components most likely associated with these effects will be evaluated in diet sensitive (C57BL/6) and genetic models of Type II diabetes susceptibility (KKA/y). In Aim 2, we will investigate the effect of co-exposure of multipollutant CAP with ozone on the temporal development of insulin resistance and adipose/lung inflammation using the KKA/y model. We will assess dose response relationship of multipollutant-O3 mixture on insulin resistance measures (HOMA-IR and IPGTT) and novel mediators of innate immune, pivotal in the development of metabolic derangement. Based on data from Aims 1 and 2, we will design experiments in Aim 3, which will help us assess chronic effects of multipollutant CAP in potentiating inflammatory monocyte activation and infiltration into tissue niches as a central mechanism for mediating adverse metabolic effects of CAP. Using state of the art multiple exposure systems available at OSU (OASIS-1 and OASIS-2) and MI in conjunction the resources available at the ECC including the use of several novel and novel high-time resolution exposure characterization methods, GLACIER offers an unprecedented opportunity to elucidate relevant mechanisms responsible for the effects of multipollutant CAP on the pathogenesis of insulin resistance and inflammation. The insights gleaned from the acute studies planned in Projects 1 and 2 in conjunction with chronic studies in Project 3, have significant public health ramifications and may eventually lead to policy changes to avert environmental exposure to PM2.5.
Progress Summary:
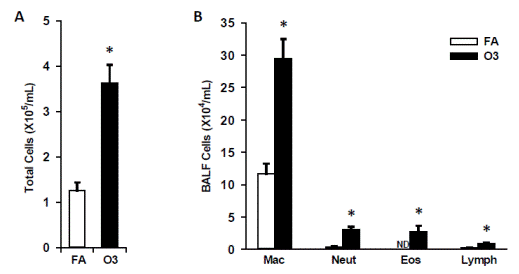
In our prior year we successfully conducted experiments to determine the temporal course of diabetes development in response to ozone and reported these results in several manuscripts- in collaboration with Dr. Harkema. We were interested in isolating the effects of ozone prior to conducting multi-pollutant exposures in combination with CAPS which we had proposed performing as part of Aim 3. We hypothesized that ozone induces rapid effects on the cardiovascular system and proceeded to investigate this in a murine model of Type II diabetes. Inhalation exposure to O3 was conducted in whole-body exposure chambers, and KK mice were exposed to nominal ozone concentrations of 0 (filtered room air), or 0.5 ppm, for 8 h/d, 7 d/wk for 13 wk (n = 8/group). Ozone was generated with two OREC Model OZONEV1-O ozonizers (Ozone Research and Equipment Corp., Phoenix, AZ), with compressed air used as a source of oxygen. The concentration of ozone within the chambers was monitored throughout the exposure with three Dasibi 1003 AH ambient-air ozone monitors (Dasibi Environmental Corp., Glendale, CA). The air-sampling probes were placed in the breathing zone of the mice. The chamber ozone concentration was automatically maintained through a computer-controlled closed-loop feedback system, which adjusted the amount of ozone delivered to the chamber through remotely controlled mass-flow valves. After three weeks of inhalation exposure to control air or ozone, mice were sacrificed. The weights of whole body and organs were recorded and metabolic as well as inflammation were examined. FIGURES 1 and 2 depict the effect of ozone exposure on lung inflammation. The total inflammatory cells in the bronchoalveolar lavage fluid (BALF) increased by ~3-fold (FIGURE 1A) and cellular component analysis suggested macrophage, neutrophil, eosinophil, and lymphocytes all increased (FIGURE 1B).
| FIGURE 1 Effect of ozone exposure on bronchoalveolar cellularity. Total cells (A) and inflammatory cell differentials (B) were enumerated in bronchoalveolar lavage fluid as described in Methods. ND - not detected; * indicates significantly different from filtered air (FA) exposed mice; p<0.05.
|
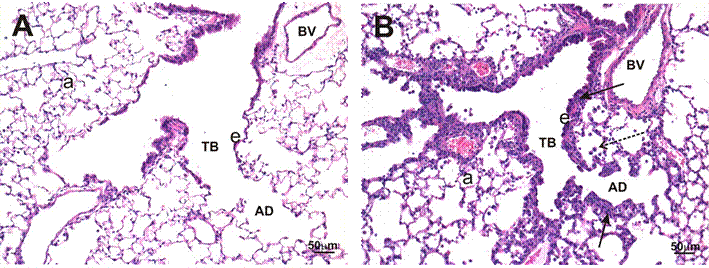
Histological examination confirmed the infiltration of immune cells in the airway. Ozone-exposed KK mice had pulmonary histopathology that was restricted to centriacinar regions throughout the lung lobe compared to FA-exposed mice (FIGURE 2A & 2B). These ozone-induced lung lesions were morphologically characterized by marked thickening of terminal bronchioles and proximal alveolar ducts as a result of hyperplasia/hypertrophy of surface epithelium, intramural fibrosis, smooth muscle hypertrophy and a mixed inflammatory cell infiltrate (i.e., mononuclear cells, neutrophils and eosinophils; bronchiolitis/alveolitis). Conspicuous aggregates of monocytes/macrophages and lesser numbers of other inflammatory cells (i.e., neutrophils, eosinophils, and lymphocytes) were also present in centrinacinar airspaces. No exposure-related histopathology was found in the lungs of KK mice exposed only to filtered air (FIGURE 2A). These results are depicted in FIGURE 2.
|
FIGURE 2 Effect of ozone exposure on pulmonary histopathology. Light photomicrographs of lung tissue from KK mice exposed to filtered air (A) or 0.5 ppm ozone (B). No exposure-related lung lesions are present in the centriacinar region of the lung from a control mouse exposed only to filtered air (A). In contrast, airway walls (solid arrows) of terminal bronchioles (TB) and alveolar ducts (AD), in the centriacinus of the lung, from an ozone-exposed mouse (B) are markedly thickened due to hyperplasia/hypertrophy of surface epithelium (e), intramural fibrosis, smooth muscle hypertrophy, and an influx of mixed inflammatory cells. Small aggregates of monocytes/macrophages (stippled arrow) are also present in alveolar airspaces in B. a, alveolar parenchyma. Tissue sections were stained with hematoxylin and eosin. |
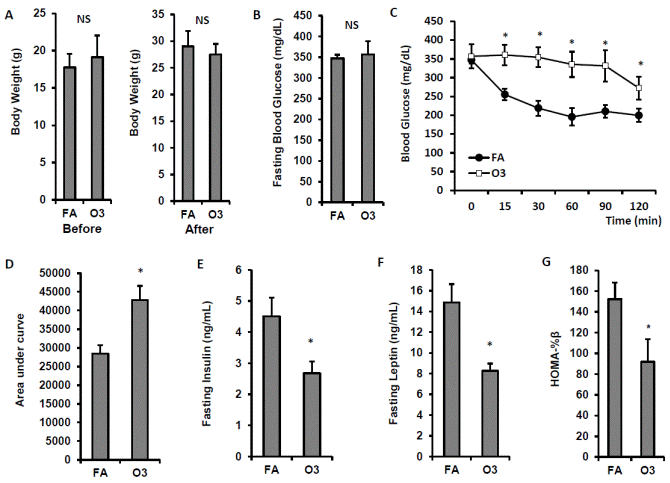
After 3 weeks of exposure, there were no differences in body weight between the O3- and FA-exposed animals (FIGURE 3A). Although no significant difference in fasting blood glucose was observed between FA- and O3-exposed animals (FIGURE 3B), three-week O3 exposure significantly impaired insulin sensitivity in KK mice (FIGURE 3C & 3D, p<0.05). Fasting plasma insulin levels were lower in O3-exposed mice (4.5 ± 0.6 ng/mL vs. 2.7 ± 0.4 ng/mL for FA vs. O3, p<0.05, FIGURE 3E). Consistently, fasting plasma leptin level and HOMA-beta were also reduced after O3 exposure in KK mice (FIGURE 3F & 3G).
| FIGURE 3 O3 exposure impaired insulin sensitivity. KK mice were exposed to O3 or FA for 13 days. Body weight was measured before and after exposure (A). ITT was performed 1 day after the last exposure. Blood glucose level was detected before and 0, 15, 30, 60, 90, 120 min after i.p. injection of 0.75U/kg body weight insulin (B, fasting blood glucose; C, blood glucose level upon insulin injection; D, area under curve). Plasma was used for the detection of insulin (E) and leptin (F). n=8; *, p<0.05. |
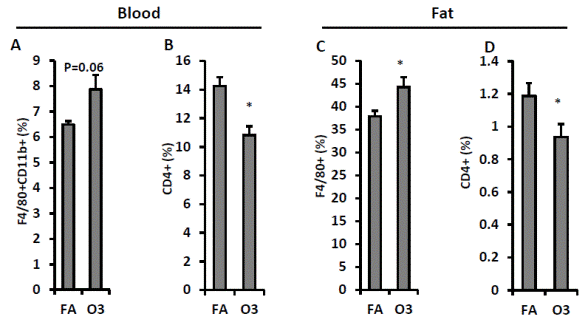
To investigate the impact of O3 inhalation on immune response, macrophage and CD4+ T cell population were quantified using flow cytometry. Monocytes were characterized by the expression CD11b and F4/80 in mice (Geissmann et al. 2003). As depicted in FIGURE 4A, CD11b+ F4/80+ monocytes increased in blood (6.5 ± 0.1% vs. 7.9 ± 0.6% for FA vs. O3) although no statistical significance was observed (p=0.06). CD4+ T cell percentage in blood was lower in O3-exposed mice (14.3 ± 0.6% vs. 10.8 ± 0.6% for FA vs. O3, p=0.001, FIGURE 4B). Consistently, infiltration of adipose tissue macrophage (ATM) increased in epididymal fat of KK mice exposed to O3 (37.9 ± 1.2% vs. 44.3 ± 2.1% for FA vs. O3, p=0.02, FIGURE 4C) and percentage of CD4+ T cell decreased in epididyma fat of O3-exposed KK mice (1.2 ± 0.08% vs. 0.9 ± 0.08% for FA vs. O3, p=0.04, FIGURE 4D).
|
FIGURE 4 Effect of O3 exposure on macrophages and CD4+ T cells. White blood cells isolated from peripheral blood and SVF harvested from epididymal fat were used for the flow cytometric detection of macrophage and T cell population. Percentage of macrophage in blood (F4/80+ CD11b+, A) and epididymal fat (F4/80+, C) was shown.CD4+ T cells in blood (B) and epididymal fat (D) were also detected. n=8; *, p<0.05. |
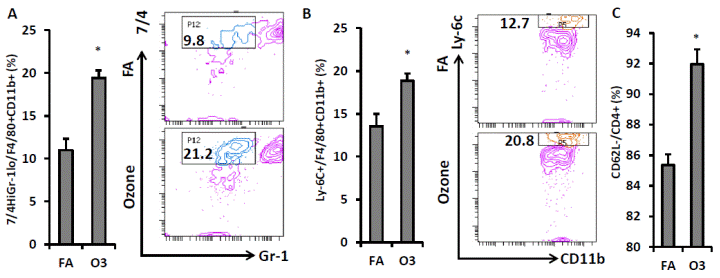
Next we detected the activation of monocytes and CD4+ T cells in the blood of O3-exposed KK mice. Gr-1low 7/4high inflammatory monocytes were significantly increased in the circulation of KK mice exposed to O3 (11.0 ± 1.3% vs. 19.4 ± 0.9% for FA vs. O3, p<0.001, FIGURE 5A). To confirm this result, we used another marker Ly-6C to identify inflammatory monocytes. Consistently, Ly-6C+ monocyte level in circulation was higher in O3-exposed mice compared with that in FA-exposed mice (13.6 ± 1.4% vs. 18.9 ± 0.8% for FA vs. O3, p=0.006, FIGURE 5B). The activated CD4+ T cells (CD4+ CD62L-) also increased after O3 exposure (85.4 ± 0.7% vs. 92.0 ± 1.0% for FA vs. O3, p<0.001, FIGURE 5C).
| FIGURE 5 Impact of O3 exposure on activation of circulating macrophages and CD4+ T cells. White blood cells isolated from peripheral blood were used for the flow cytometric detection of macrophage and T cell activation. F4/80+ CD11b+ macrophages were gated for the analysis of inflammatory marker. Inflammatory macrophages as evidenced by 7/4high Gr-1low (A) or Ly-6C+ (B) were analyzed (Left, statistical analysis; Right, representative images). CD4+ cells were gated for the detection of T cell activation. Activated CD4+ T cells (CD4+ CD62L-) were shown (C). n=8; *, p<0.05. |
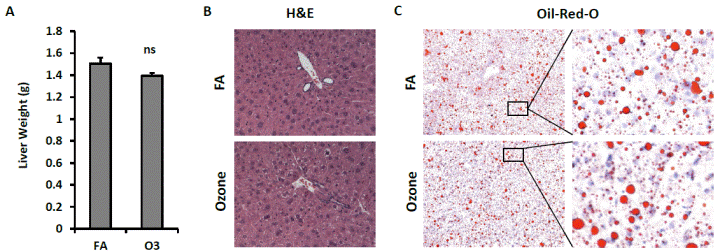
As depicted in FIGURE 6A, KK mice exposed to O3 and FA had similar liver weights. H&E staining shows normal liver architecture and morphology in mice exposed to O3 (FIGURE 6B). To investigate whether O3 exposure affects hepatic lipid content, liver frozen sections were used for Oil-Red-O staining. As shown in FIGURE 6C, similar levels of lipid content were observed in liver of mice exposed to FA and O3.
| FIGURE 6 Effect of O3 exposure on liver weights and lipid content. A, Liver weight of FA- or O3-exposed mice; B, Sections of liver were used for the H&E staining and representative images were shown; C, Oil-Red-O staining of frozen liver sections. |
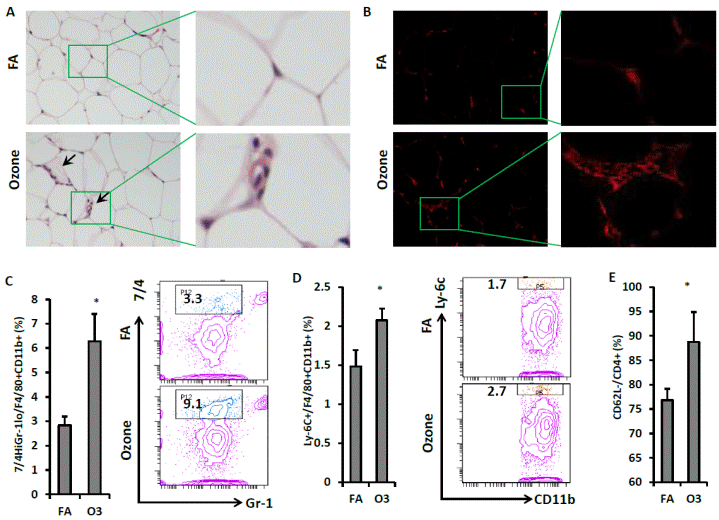
To investigate inflammatory cell infiltration in visceral adipose tissue, epididymal fat tissues from exposed mice were used for the H&E staining and Mac-1 immunofluorescence staining. More crown-like structures (infiltration of inflammatory cells) were observed in the epididymal fat of O3-exposed mice (FIGURE 7A & 7B). Consistent with the observations in blood, infiltration of inflammatory macrophages in visceral adipose tissue was enhanced by O3 exposure. As shown in FIGURE 7C, percentage of Gr-1low 7/4high macrophage increased in the epididymal fat of O3-exposed mice (2.8 ± 0.4% vs. 6.3 ± 1.1% for FA vs. O3, p=0.02,). Ly-6C+ macrophage infiltration in the epididymal fat of O3-exposed mice is also higher than that of mice exposed to FA (1.5 ± 0.2% vs. 2.1 ± 0.2% for FA vs. O3, p=0.04, FIGURE 7D). There was also an increased activation of CD4+ T cell in the adipose tissue of O3-exposed KK mice (76.8 ± 2.3% vs. 88.8 ± 6.1% for FA vs. O3, p<0.001, FIGURE 7E).
| FIGURE 7 O3 exposure promoted activation of macrophages and CD4+ T cells in visceral adipose tissue. Sections of epididymal fat were used for the H&E staining (A) and immunofluorescence staining (B, CD11b). Representative images were shown (arrows indicate crown-like structure infiltrated by inflammatory cells/macrophages). SVF harvested from epididymal fat was used for the flow cytometric detection of macrophage and T cell activation. F4/80+ macrophages were gated for the analysis of inflammatory marker. Inflammatory macrophages as evidenced by 7/4high Gr-1low (C) or Ly-6C+ (D) were analyzed (Left, statistical analysis; Right, representative images). CD4+ cells were gated for the detection of T cell activation. Activated CD4+ T cells (CD4+ CD62L-) were shown (E). n=8; *, p<0.05. |
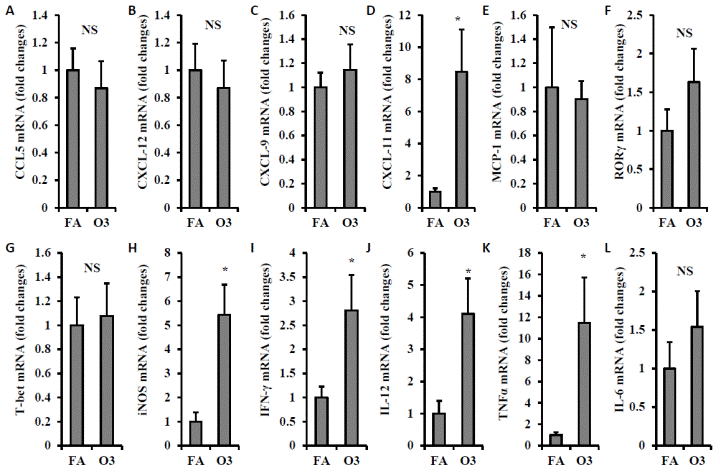
To further investigate the impact of ozone inhalation on immune activation in adipose tissue, we next detected the mRNA expression level of chemokines, pro-inflammatory cytokines, and inflammatory genes in visceral adipose tissue. No significant difference in CCL-5, CXCL-12, CXCL-9, MCP-1, RORγ, T-bet and IL-6 were observed between FA- and O3-exposed mice (FIGURE 8A-C, 8E-G, and 8L). Expression of CXCL-11 was increased by 8.5-fold in animals exposed to O3 (1.0 ± 0.22 vs. 8.5 ± 2.63 for FA vs. O3, p=0.02, FIGURE 8D). There were also a 5.4-fold increase of iNOS (1.0 ± 0.39 vs. 5.4 ± 1.26 for FA vs. O3, p=0.007, FIGURE 8H), 2.8-fold increase of IFN-γ (1.0 ± 0.23 vs. 2.8 ± 0.73 for FA vs. O3, p=0.045, FIGURE 8I), 4.1-fold increase of IL-12 (1.0 ± 0.40 vs. 4.1 ± 1.10 for FA vs. O3, p=0.026, FIGURE 8J), and 11.5-fold increase of TNFα (1.0 ± 0.24 vs. 11.5 ± 4.24 for FA vs. O3, p=0.039, FIGURE 8K).
| FIGURE 8 Effect of ozone exposure on inflammatory gene expression in visceral adipose tissue. Epididymal fat of mice exposed to filtered air (FA) or ozone (O3) was used for real-time PCR detection of inflammatory gene expression. mRNA expression of chemokines(A, CCL-5; B, CXCL-12; C, CXCL-9; D, CXCL-11; E, MCP-1), inflammatory genes (F, RORγ; G, T-bet; H, iNOS), and inflammatory cytokines (I, IFN-γ; J, IL-12; K, TNFα; L, IL-6) was shown . N=8/group; *, p<0.05. |
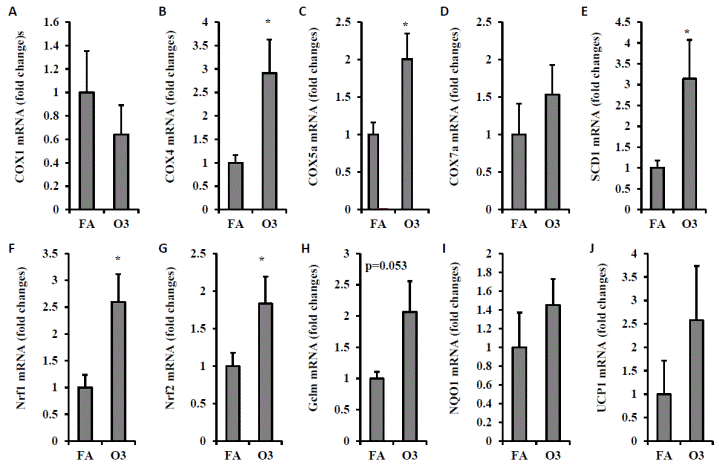
To investigate whether oxidative stress plays a role in ozone exposure, we detected the expression of genes involved in oxidative stress. As depicted in FIGURE 9, multiple oxidative stress-related genes including COX4 (1.0 ± 0.16 vs. 2.9 ± 0.71 for FA vs. O3, p=0.021, FIGURE 9B), COX5α (1.0 ± 0.16 vs. 2.0 ± 0.34 for FA vs. O3, p=0.016, FIGURE 9C), SCD1 (1.0 ± 0.18 vs. 3.1 ± 0.93 for FA vs. O3, p=0.041, FIGURE 9E), Nrf1 (1.0 ± 0.24 vs. 2.6 ± 0.52 for FA vs. O3, p=0.014, FIGURE 9F), and Nrf2 (1.0 ± 0.17 vs. 1.8 ± 0.36 for FA vs. O3, p=0.045, FIGURE 9G) were up-regulated by the exposure of O3.
| FIGURE 9 Ozone exposure induces expression of cyclooxygenase (COX) and oxidative stress response in VAT. Epididymal fat of mice exposed to filtered air FA or O3 was used for real-time PCR detection of genes. A, COX1; B, COX4; C, COX5a; D, COX7a; E, SCD1; F, Nrf1; G Nrf2; H, Gclm; I, NOQ1; J, UCP1. N=8/group; *, p<0.05. |
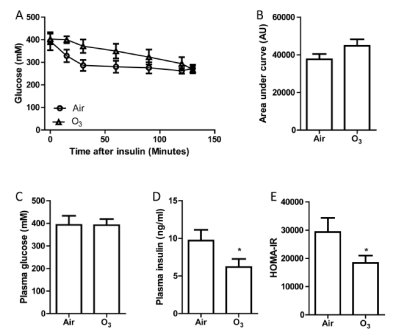
We next detect the effect of ozone exposure on KKAy, a type 2 diabetes model with severe hyperinsulinemia. Interestingly, ozone exposure increased insulin sensitivity in KKAy mice unlike that in KK mice (FIGURE 10). This result suggests the effect of ozone on insulin resistance might be genetic background-specific.
| FIGURE 10. Ozone inhalation increases insulin sensitivity in KKAy mice. KKAy mice were exposed to air or ozone for 13 days. After 6-hour fasting (2 hours after the last exposure), these mice were subjected to ITT. A, the plasma glucose levels at different time points after intraperitoneal injection of insulin. B, the area under curve of ITT. C, fasting blood glucose levels. D, fasting plasma insulin levels. E, HOMA-IR. *p<0.05 vs Air, student’s t test. n=8/group. |
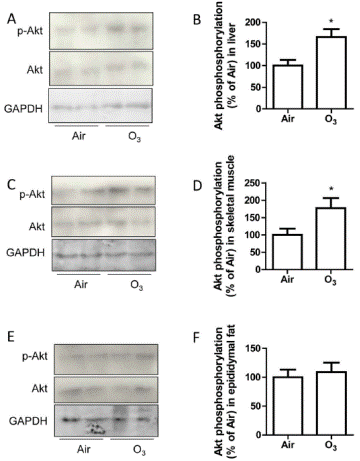
Then insulin signal pathways were detected in the KKAy mice that had been exposed to FA or ozone. The phosphorylation of Akt increased in skeletal muscle and liver, but not adipose tissues isolated from KKAy mice exposed to ozone (FIGURE 11).
| FIGURE 11. Ozone inhalation enhances insulin-induced Akt phosphorylation in tissues. KKAy mice were exposed to air or ozone for 13 days, and liver, skeletal muscle, and epididymal fat tissues were collected and treated with insulin (0.5 U/ml) for 5 minutes. Akt ser473 phosphorylation levels in liver (A and B), skeletal muscle (C and D), and epididymal adipose tissues (E and F) were analysed by western blot. *p<0.05 vs Air, student’s t test. n=8/group. |
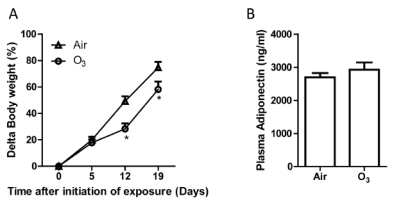
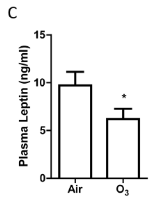
The body weight significantly reduced in ozone-inhaled KKAy mice compared to that exposed to FA (FIGURE 12A). The plasma level of leptin but not adiponectin reduced in ozone-inhaled KKAy mice (FIGURE 12B & 12C), suggesting weight loss and leptin sensitization may be associated with the increased insulin sensitivity in KKAy mice exposed to onzone.
|
FIGURE 12. Ozone inhalation reduces weight gain and decreases plasma leptin levels. The body weights of Air- and ozone-exposed KKAy mice were measured weekly, and the body weight gain is presented. *p<0.05 vs Air, two way ANOVA. n=8/group. B and C: After 13 days of inhalation exposure to ozone or air, KKAy mice were euthanized, and the levels of adiponectin (B) and leptin (C) were assessed by ELISA. *p<0.05 vs Air, student’s t test. n=8/group. |
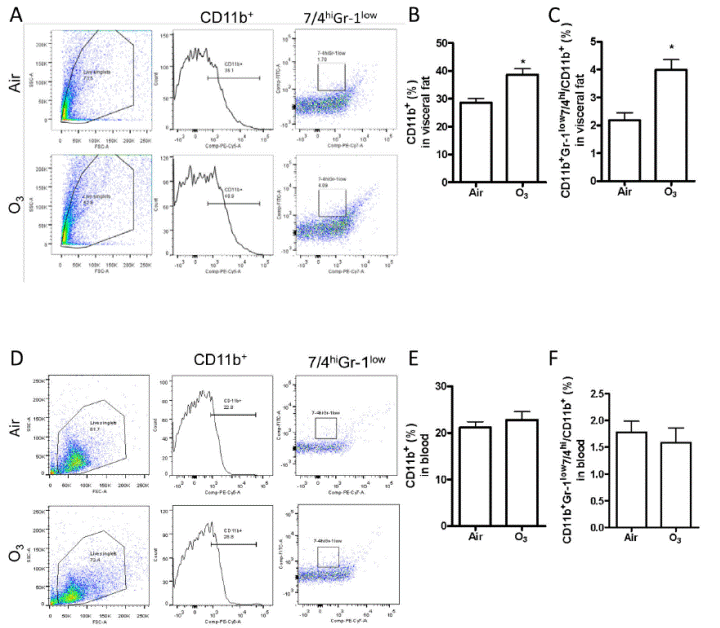
We then detected the level of inflammation in the adipose tissue. As depicted in FIGURE 13A-C, macrophage number and macrophage activation increased in the adipose tissue of ozone-inhaled animals. In contrast, the macrophage number and macrophage activation did not changed in the peripheral blood (FIGURE 13D-F).
| FIGURE 13. Ozone inhalation induces inflammation in adipose tissues. After exposure to air or ozone for 13 days, macrophage infiltration and activation in response to ozone exposure was assessed by flow cytometry. Cells were isolated from epididymal adipose tissues (A–C) and blood (D–F). Both representative plots of flow cytometric analysis (A and C) and the richness of total (CD11b+, B and E) and proinflammatory (CD11b+Gr-1low7/4hi, C and F) macrophages are presented. *p<0.05 vs Air, student’s t test. n=8/group. |
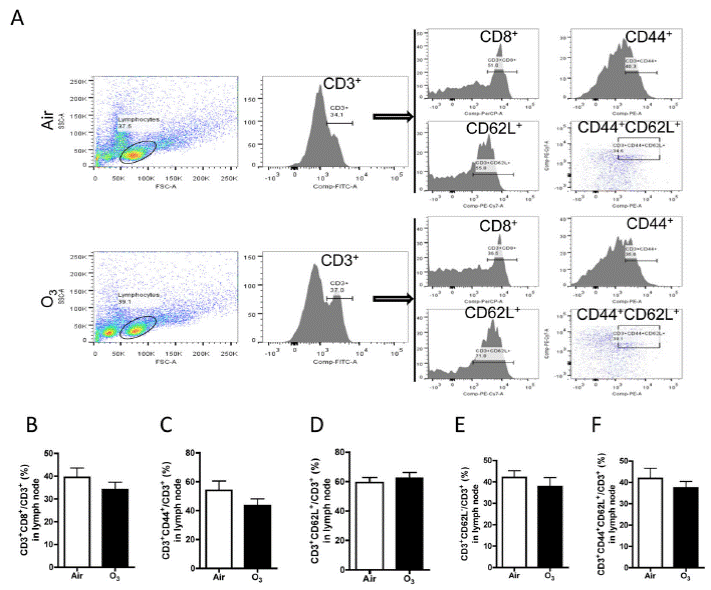
Next, we also examined the activation of T cells in the mediastinal lymph nodes. As shown in FIGURE 14, there was no significant difference in T cell activation in both groups.
| FIGURE 14. Ozone inhalation does not change the profile of T cell subsets. After exposure to air or ozone for 13 days, mediastinal lymph nodes were collected, and T cell subsets were profiled with flow cytometry. A, representative plots of flow cytometric analysis. B–F, the richness of T cell subsets defined by the expression of indicated surface markers. |
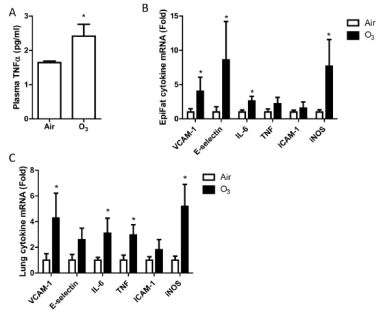
At last, we detected inflammatory mediators in the blood, adipose tissue, and lung. Plasma TNFa level significantly increased after ozone exposure (FIGURE 15A). The expressions of VCAM-1, E-selectin, IL-6, and iNOS were higher in the epididymal fat from ozone-inhaled animals compared to those exposed to FA (FIGURE 15B). Similarly, there were higher levels of VCAM-1, IL-6, TNFa, and iNOS in the lung of ozone-exposed mice (FIGURE 15C).
| FIGURE 15. Ozone inhalation induces extra-pulmonary inflammation. After exposure to air or ozone for 13 days, pro-inflammatory cytokine levels in blood, epididymal fat tissues, and lung were assessed. A, plasma TNFα protein levels, measured by ELISA. *p<0.05 vs Air, student’s t test. n=8/group. B and C, pro-inflammatory gene mRNA expression in epididymal fat tissues (B) and lung (C), assessed by real-time RT-PCR. *p<0.05 vs Air, one way ANOVA. n=8/group. |
Future Activities:
All aspects of the study protocol are approved by our IACUC. We anticipate beginning our CAPS + Ozone exposure at the end of this year. We plan to expose 8-week-old C57BL/6 mice on normal chow or high fat diet to filtered air or CAPS + ozone (10 mice/group) for 8 weeks. The effects on insulin sensitivity, oxidative stress, and inflammation will be examined.
Journal Articles on this Report : 2 Displayed | Download in RIS Format
| Other subproject views: | All 45 publications | 34 publications in selected types | All 34 journal articles |
|---|---|---|---|
| Other center views: | All 148 publications | 72 publications in selected types | All 72 journal articles |
| Type | Citation | ||
|---|---|---|---|
|
|
Subacute inhalation exposure to ozone induces systemic inflammation but not insulin resistance in a diabetic mouse model. Ying Z, Allen K, Zhong J, Chen M, Williams KM, Wagner JG, Lewandowski R, Sun Q, Rajagopalan S, Harkema JR. Inhal Toxicol. 2016;28(4):155-63. |
R834797C003 (2016) R834797C003 (Final) |
not available |
|
|
Repeated ozone exposure exacerbates insulin resistance and activates innate immune response in genetically susceptible mice. Zhong J, Allen K, Rao X, Ying Z, Braunstein Z, Kankanala SR, Xia C, Wang X, Bramble LA, Wagner JG, Lewandowski R, Sun Q, Harkema JR, Rajagopalan S. Inhal Toxicol. 2016 Aug;28(9):383-92. |
R834797C003 (2016) R834797C003 (Final) |
not available |
Supplemental Keywords:
Ozone (O3) PM2.5, Type II Diabetes Mellitus (DM), Insulin Resistance (IR), Air, Scientific Discipline, ENVIRONMENTAL MANAGEMENT, Health Risk Assessment, Biology, Biochemistry, air toxics, Risk Assessment, aerosol particles, physiology, human exposure, particulate matter, cardiotoxicity, susceptible populations, acute cardiovascualr effects, ambient air quality, cardiopulmonaryProgress and Final Reports:
Original AbstractMain Center Abstract and Reports:
R834797 Center for Research on Early Childhood Exposure and Development in Puerto Rico Subprojects under this Center: (EPA does not fund or establish subprojects; EPA awards and manages the overall grant for this center).
R834797C001 Cardiometabolic Effects of Exposure to Differing Mixtures and Concentrations of PM2.5 in Obese and Lean Adults
R834797C002 Cardiometabolic, Autonomic, and Airway Toxicity of Acute Exposures to PM2.5 from Multipollutant Atmospheres in the Great Lakes Region
R834797C003 Long Term Metabolic Consequences of Exposures to Multipollutant Atmospheres in the Great Lakes Region
The perspectives, information and conclusions conveyed in research project abstracts, progress reports, final reports, journal abstracts and journal publications convey the viewpoints of the principal investigator and may not represent the views and policies of ORD and EPA. Conclusions drawn by the principal investigators have not been reviewed by the Agency.