
Grantee Research Project Results
2008 Progress Report: Quantitative Assessment of Pathogens in Drinking Water
EPA Grant Number: R833002Title: Quantitative Assessment of Pathogens in Drinking Water
Investigators: Schwab, Kellogg J. , Halden, Rolf U. , Graczyk, Thaddeus
Institution: The Johns Hopkins University
EPA Project Officer: Aja, Hayley
Project Period: August 25, 2006 through August 24, 2009 (Extended to September 30, 2010)
Project Period Covered by this Report: August 25, 2007 through August 24,2008
Project Amount: $600,000
RFA: Development and Evaluation of Innovative Approaches for the Quantitative Assessment of Pathogens in Drinking Water (2005) RFA Text | Recipients Lists
Research Category: Drinking Water , Water
Objective:
A major limiting factor in assessing the human health risk of microbial pathogens in raw and finished drinking water is the lack of robust, efficient methods for concentrating, identifying and quantifying low levels of bacteria, viruses and protozoa simultaneously, effectively and rapidly. We will develop a microbial isolation and detection protocol capable of qualitative and quantitative identification of waterborne microbial pathogens by combining the latest high-efficiency filtration technology with rapid and sensitive molecular detection techniques including quantitative PCR (qPCR), quantitative reverse transcription-PCR (qRT-PCR), fluorescent in situ hybridization (FISH) and liquid chromatography tandem mass spectrometry (LC-MS/MS). The sensitivity and specificity of the proposed pathogen recovery and detection approach will be directly compared to current USEPA methods via spiking and analysis of raw and finished drinking water samples collected from various water resources and distribution systems. Following method validation, a series of unspiked raw or finished waters (including waters from distribution systems) will be monitored for pathogenic microorganisms to demonstrate the utility of the approach in real world situations.
Progress Summary:
Research during the second year (August 2007-August 2008) of this project included efforts to determine the detection threshold of the multiplexed FISH assay for oocysts of C. parvum and cysts of G. lamblia; and the detection threshold of the multiplexed FISH assay for spores of human-virulent microsporidia (i.e., E. bieneusi, E. intestinalis, E. hellem, and E. cuniculi). In addition, we investigated whether we could combine the FISH assay for Cryptosporidium and Giardia with the FISH assay for microsporidia into a single multiplexed FISH assay. Progress has also been made towards the further development of mass spectrometry (MS) methods for the detection and quantification of virus capsid proteins in complex matrixes containing non-target microbial proteins. More specifically, liquid chromatography tandem MS (LC-MS/MS) has been applied to the detection of virus-like particles (VLPs) within complex samples containing Escherichia coli K12 proteins. Currently, the sample preparation method is being optimized in order to reduce its complexity and to improve the sensitivity of the assay. In addition, a tangential flow ultrafiltration system has been further optimized for simultaneously recover of viruses, spore-forming bacteria, and vegetative bacteria from 100L water samples of varying type and quality. A series of commercially available hollow fiber membranes have been compared, and one has been selected for further application based on performance and cost. Molecular detection methods have been developed and applied for the detection of murine norovirus (MNV-1) in select sample concentrates. In addition, research in the development of methods for post-ultrafiltration concentration and detection of these recovered microorganisms is ongoing. A summary of the research conducted during the current reporting period (August 2007-August 2008) is presented below.
Microbial Stock Production and Detection Methods: The recovery and downstream detection of several microorganisms have been developed in laboratory studies evaluating the tangential flow ultrafiltration system. During the ongoing optimization of FISH assays and the mass spectrometry method, select microorganisms were utilized. In addition, US EPA approved methods for the detection of indicator organisms (i.e. total coliforms, Escherichia coli, and Enterococcus) are currently being utilized in the analysis of select environmental samples. The previous annual report described the propagation and detection of MS2 bacteriophage, E. coli CN-13, enteric viruses (i.e. MNV-1), Cryptosporidium parvum oocysts, Giardia lamblia cysts, and spores of human-virulent microsporidia including E. intestinalis, E. hellem, E. cuniculi, and E. bieneus. Additional microorganisms have been added over the past 12 months all of which have been used extensively in our laboratories and can be successfully propagated and analyzed by conventional diagnostic assays as described in Appendix 1.
Detection of C. parvum oocysts, G. lamblia cysts, and microsporidian spores in spiked water samples using multiplexed FISH assays: A series of experiments were carried out in which 100 ml water samples obtained from the processing of 10 L of drinking water samples according to the US EPA Method 1623 were spiked with 10 C. parvum oocysts and 10 G. lamblia cysts. The 10 L drinking water samples were pre-filtered using 1.2-μm-pore size cellulose acetate membrane. The water samples (100 ml) were processed using multiplexed FISH assay to identify oocysts and cysts. In all experiments where 10 C. parvum oocysts and 10 G. lamblia cysts were used as inoculum, the oocysts and cysts were detectable by the multiplexed FISH assay. However, spiking of 100 ml of water samples with less than 10 oocysts and cysts produced negative results. A similar series of experiments were carried out for microsporidian spores. However, spiking of 100 ml of water with 10 spores of E. bieneusi, E. intestinalis, E. hellem, and E. cuniculi produced negative results.
An additional series of experiments were carried out to combine these two aforementioned multiplexed FISH assays into a single FISH assay. Although microsporidian spores were clearly visible in material processed by the FISH, all experiments produced negative results for C. parvum oocysts and G. lamblia cysts. It was concluded that the incubation time of 3 hrs at 57°C has diminished binding capacity of anti-C. parvum and anti-G. lamblia monoclonal antibodies. All FISH reactions were carried out as described in the previous progress report (2006-2007) in the section on detection methods.
Mass Spectrometric Detection of Virus-like Particles: Incorporating recent advances in the field of mass spectrometry, we have moved from matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) to liquid chromatography tandem mass spectrometry (LC-MS/MS) coupled with selective reaction monitoring for absolute quantification of three viral peptide biomarkers originating from the capsid protein of our model norovirus (AQUA approach). This method uses an internal standard technique commonly applied in environmental small molecule analysis. In order to demonstrate that the AQUA method is applicable beyond purified standards (where we were able to detect viral proteins as low as 5 x 10-16 moles) we challenged the method by spiking 100 femtomoles (1 fmol = 10-15 moles) of virus-like particles (VLPs) into 15 μg of a complex matrix of Escherichia coli K12 proteins. As outlined in Figure 1, VLP-spiked E. coli cell lysates were separated by SDS-PAGE and visualized with Coomassie colloidal blue stain.
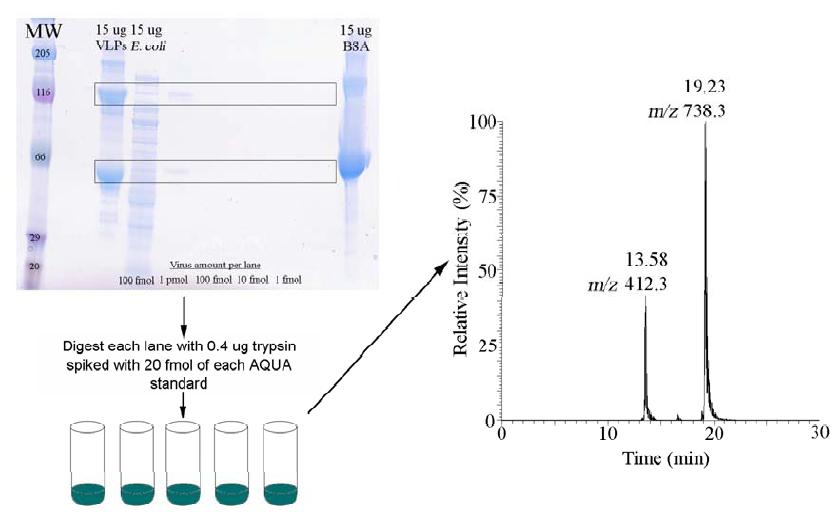
Figure 1. Illustration of the experimental strategy developed for virus identification and quantification in samples using AQUA standards. Spin column-purified samples are separated using gel electrophoresis. Bands of interest are then excised and gel pieces spiked with AQUA standards and digested with trypsin. Recovered digests are then detected using liquid chromatography tandem mass spectrometry.
Using purified VLPs as a molecular weight standard, regions of the gel containing the capsid proteins were excised and digested using standard protocols. Internal standards were spiked in prior to digestion. Tryptic peptides were then extracted and analyzed by LC-MS/MS on an LCQ Deca XP (Thermo Finnigan) ion trap mass spectrometer. Calculating concentration based upon peak area, we measured 79 ± 14.6 femtomoles of protein in the samples (Figure 2). This suggests that there are some losses in the sample preparation, and that reported values are likely underestimating to the amount of virus present in the sample. Using a lower spike of VLPs (10 fmol) in 15 μg of the same E. coli, the recovery was measured as 54 ± 63%. The high error around this value reflects the challenge of detecting very low amounts of target in a complex matrix. The AQUA method, which specifically targets our ions of interest, provides an increase in sensitivity. Full scans of the sample yielded only identification of E. coli proteins, and did not find the viral peptides of interest, where the AQUA approach measured the specific ion transitions unique to the viral capsid proteins. Thus the AQUA system shows significant promise with respect to detection of target microorganisms in complex matrixes.
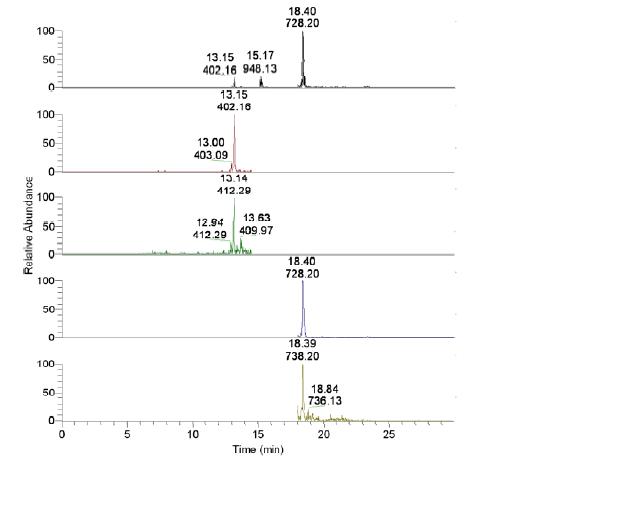
Figure 2. Target peptides were recovered following spiking into 15 μg of Escherichia coli crude cell lysate at 100 fmol. The average calculated concentration was 79 ± 14.6 femtomoles of target peptides. Labels show retention time and m/z ratio, respectively. The m/z 412 and 738 represent internal standards; the m/z 402 and 728 are markers of the viral peptides.
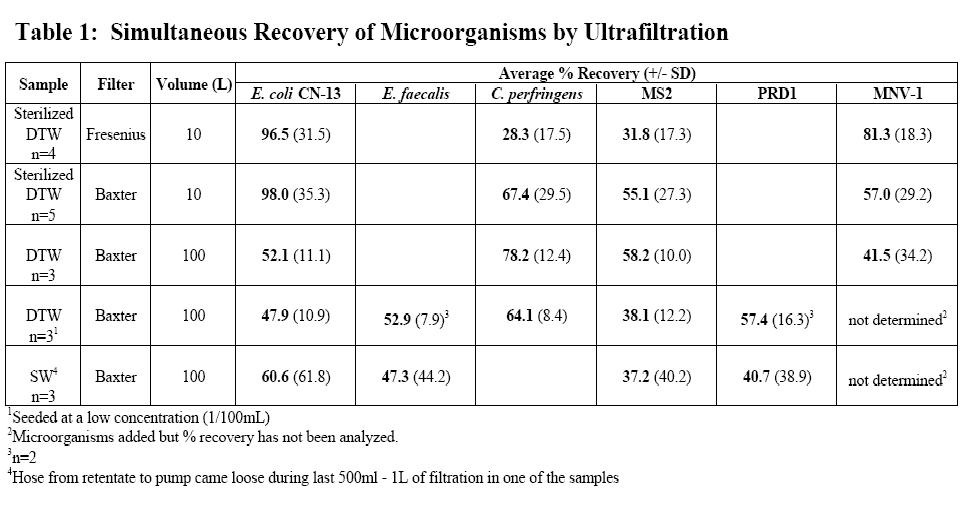
Lab-based Ultrafiltration Experiments: Optimization of the tangential flow ultrafiltration system has progressed during the second reporting period. In the previous reporting period (August 25, 2006 through August 24, 2007), we evaluated the ability of a capsule filter, available through Fresenius Medical Care (Waltham, MA), to simultaneously concentrate MS2 bacteriophage, E. coli CN-13, and MNV-1 from 10L water samples (n=9). The Fresenius capsule filter contains polysulfone hollow fiber membranes with an average molecular weight cut-off (MWCO) of 30kDa and a total surface area of 1.8m2. During the current reporting period (August 25, 2007 through August 24, 2008), we compared the Fresenius capsule filter to another commercially available filter from Baxter International Inc. (Deerfield, IL). The Baxter Exceltra Plus 210 capsule filter contains cellulose triacetate hollow fiber membranes with an average MWCO of 70kDa and a total surface area of 2.1m2. Cellulose triacetate is an extremely hydrophilic material thus allowing it to be nearly non-protein binding. The Fresenius (n=4) and Baxter (n=5) filters were compared using 10L sterilized dechlorinated tap water (DTW) samples spiked with MS2 bacteriophage, E. coli CN-13, C. perfringens spores, and MNV-1. Both of these filters are disposable and are subject to one-time use during lab and field-based evaluations. These filters preformed similarly during recovery experiments and thus the Baxter Exeltra Plus 210 was chosen for continued use based on cost per unit.
We have also modified our previously described ultrafiltration method protocol to reflect some adjustments reported in related peer-reviewed publications. As described in Polacyzk et al.,2008, filters were blocked with 1L of 0.1% Sodium Polyphosphate (NaPP) immediately before the start of filtration as opposed to overnight blocking with 500 ml of a 5% bovine serum solution (1). In addition, the elution method has been modified. Instead of eluting the filter with 500 ml of an elution buffer containing 0.5% Tween 80, 0.001% Antifoam-A, and 0.01% NaPP as described previously, a modified elution buffer (0.1% Tween 80, 0.001% Antifoam-A, and 0.01% NaPP) is added to the final concentrate at a 1:9 ratio. The concentrate with elution buffer is then recirculated at 1,700 ml/min for five minutes with the filtrate ports closed and no back-pressure. The original method and the modified method are referred to as Method 1 and Method 2, respectively, in this report.
As previously described, the ultrafiltration system is operated at a flow rate of 1,700 ml/min under 7-8 lb/in2 pressure with an average flow of 800 ml/min and 900 ml/min for the permeate rate and cross-flow rate, respectively. These parameters are achieved using a digital peristaltic pump with a high-performance pump head (Cole Parmer, Vernon Hills, IL). Throughout the filtration process, quality control samples are collected and assayed along with the spiked water samples and final concentrates. In addition, tubing and fittings are decontaminated after each experiment in a 10% bleach solution, followed by a thorough rinse with DI water, and then autoclaved at 15 psi and 121°C for 15 minutes.
A summary of the data for this reporting period are shown in Table 1. These data are based on results for microbial detection assays described previously in this report and the previous report for the first 12 months of the project. A total of 18 water samples varying in volume and type of test water were analyzed during this period. Figure 3 depicts the laboratory ultrafiltration setup used to concentrate 100L samples.

Molecular Detection of MNV-1: The protocol used for the analysis of MNV-1 RNA is described in the following section. This description represents the analysis of all the target microorganism nucleic acid (both DNA and/or RNA) with the exception of the use of microorganism-specific primers and probes, adjustments to the primer and probe concentrations and variances in primer annealing temperatures. For viral RNA amplification a QuantiTect Probe RT-PCR Kit is used. Briefly, 5μl of prepared sample is added to 20 μl of the following master mix: 1× QuantiTect Probe RT-PCR Master Mix; 1× QuantiTect Reverse Transcriptase Mix; 5 U of GeneAmp RNAse inhibitor; 0.4 μM downstream primer; 0.4 μM upstream primer; 0.2 μM probe; and 4.50 μl DEPC-treated water. The following cycling conditions are used: reverse transcription (30 min; 50oC); denaturation of the reverse transcriptase and activation of the hot-start polymerase (95ºC; 15 min); and 45 cycles of template denaturation (94ºC; 15 seconds), and primer annealing and extension (60ºC; 60 seconds) in an ABI Prism 7000 or 7300 Sequence Detection
System (Foster City, CA). Positive and negative control samples are analyzed with each thermocycler run to ensure reagent and cycling efficiency.
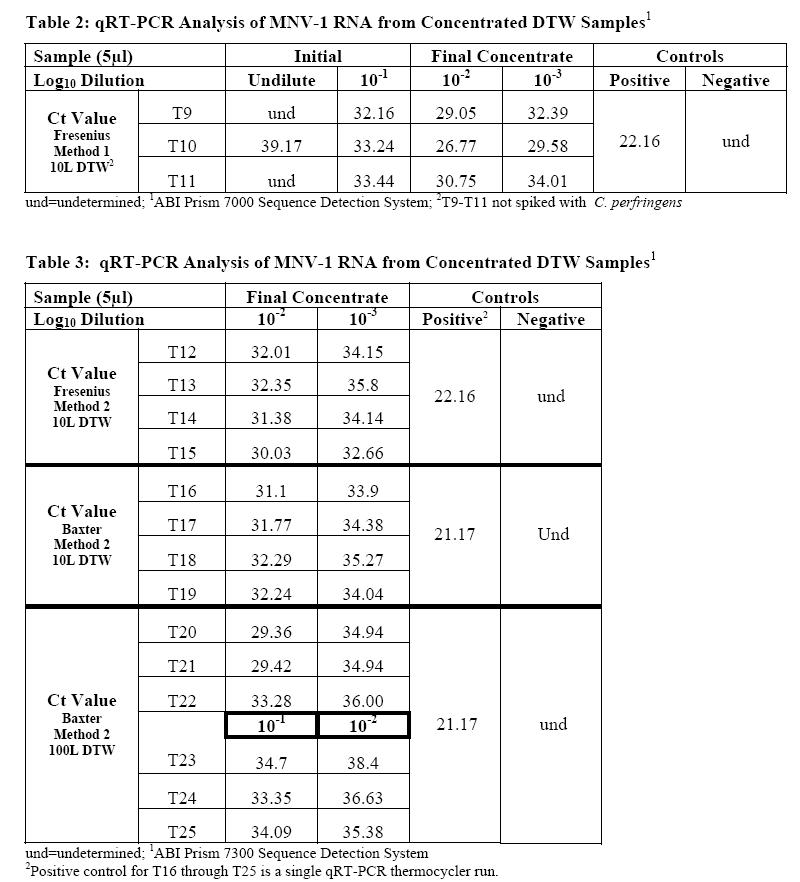
Samples from seventeen separate ultrafiltration experiments were analyzed for the presence of MNV-1. Of these experiments, seven used the Fresenius capsule filter, and ten were completed using the Baxter filter. The experiments using the Fresenius filter were 10 L tap water samples that were sterilized and dechlorinated with sodium thiosulfate. These samples (n=7) were then spiked with MNV-1, MS2, C. perfringens, and E.coli CN-13 at an average initial concentration of 4.3 x102, 2.4 x 102, 5.3 x 102, and 1.3 x 103 PFU or CFU/ml, respectively. Three of these water samples were processed using Method 1, and the remaining four were processed using Method 2. From the 3 experiments utilizing Method 1, the initial sample and final concentrated sample were analyzed for the presence of MNV-1 RNA. Samples processed using Method 2 were only analyzed for the presence of MNV-1 RNA in the final concentrate.
The Baxter filter unit was utilized in experiments with either 10L (n=4) or 100L (n=6) of DTW. The 10L DTW samples were spiked with MNV-1, MS2, C. perfringens, and E.coli CN-13 at an initial concentration of 1.3 x 102, 1.3 x 101, 3.1 x 102, and 1.6 x102 PFU or CFU/ml, respectively. The 100L DTW samples are divided into high (n=3) and lower (n=3) seed groups. The high seed group was spiked with MNV-1, MS2, C. perfringens, and E. coli CN-13 at an initial seed of 4.9 x 106, 2.3 x 106, 3.0 x 106, and 3.6 x 106 PFU or CFU/ml, respectively. The low concentration group was spiked with MNV-1, MS2, PRD1, C. perfringens, E. faecalis, and E. coli CN-13 at an initial seed of 5.5 x 104, 4.0 x 104 , 5.8 x 104, 1.35 x 105, 7.7 x 103, and 2.6 x 104 PFU or CFUs, respectively. All DTW samples using the Baxter filter were processed by Method 2.
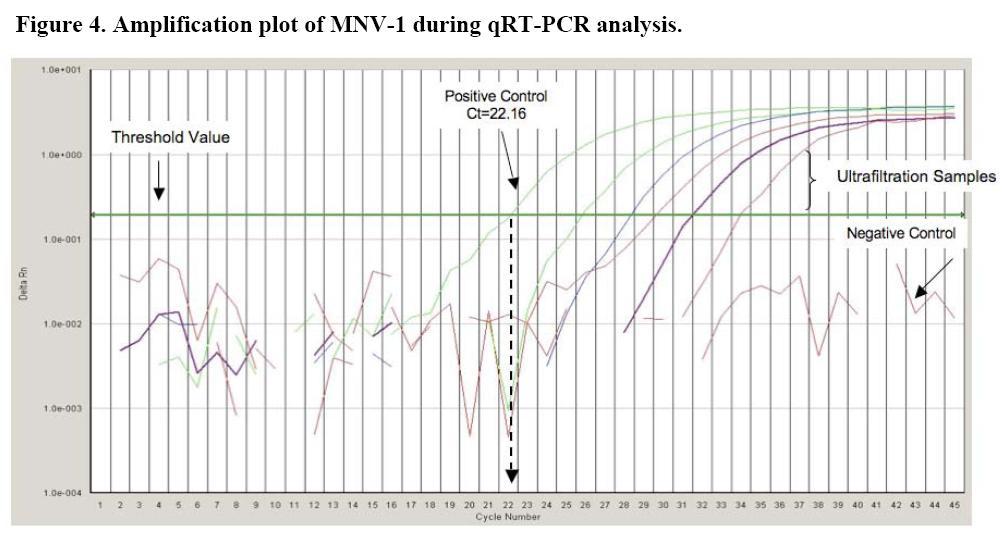
Before any of the samples were added to the prepared qRT-PCR reagent master mix, appropriate log10 dilutions were made using nuclease-free water and then the prepared samples were heat released at 95°C for 5 minutes to break open the viral protein capsid and release the RNA. Figure 4 displays the amplification plot for the qRT-PCR analysis of MNV-1. This amplification plot shows the negative and positive controls as well as typical results, or Ct values, seen with ultrafiltration samples.
Tables 2 and 3 show the results for the detection of MNV-1 using qRT-PCR. The volume of the final concentrate for these 17 experiments, or trials (T), ranged from 74 to 235 mL. Log10 dilutions were prepared from the final concentrate using DEPC-treated water, and a 5μL volume sample of the 10-2 and 10-3 dilution was analyzed for T9 through T22. For T23, T24, and T25, the 10-1 and 10-2 dilution was analyzed. The results presented in the tables indicate that the use of qRT-PCR as a tool for detection of viruses in water samples is consistent and reproducible. Target MNV-1 RNA was detected in all samples with exception to the initial undiluted sample for T9 and T11. In qRT-PCR, samples seeded with a log10 difference in microorganism concentration should have a difference of 3.3 Ct values (i.e. 3.3 cycles) (2). For T9, T10, and T11, shown in Table 2, the median slope value for MNV-1 was calculated to be -3.26 Ct/log10. For T12 through T25, the median slope value for MNV-1 was calculated to be -2.78 Ct/log10.
Appendix 1
A. PRD1 bacteriophage
1. PRD1 stock generation: The method for generation of PRD1 bacteriophage is similar to the method for generating MS2 bacteriophage previously described in the first reporting period. Briefly, to generate high titer (1011 pfu) bacteriophage stocks, a bacterial host (i.e. Salmonella typhimurium LT2) is incubated in a flask with tryptic soy broth (TSB) at 37°C with shaking for 4 hours to produce a log phase growth. The bacterial host at log phase growth is then used to propagate PRD1 using a soft agar overlay method. In this method, 75μl of prepared bacterial host and 100μl of diluted PRD1 stock are added to a sterile tube containing 5 mL of 0.7% tryptic soy agar (TSA). The contents of the tube are mixed gently and then carefully poured onto a Petri dish containing 15 mL of 1.5% TSA. These steps are repeated on a total of 4 plates. The plates are then incubated at 37°C, without inverting, for 16-18 hours. Using a cell scraper, harvest the PRD1 from the plates by gently scraping the top, soft agar layers into a 50cc tube. Next, Dulbecco’s Phosphate Buffered Saline (DPBS) is added to the 50cc tube to achieve a total volume of 23 mL followed by the addition of 23 mL of chloroform. This mixture is then vortexed for 5 minutes at maximum speed and centrifuged at 4,000xg for 30 minutes in order to separate the PRD1 from the extracellular materials and soft agar. The PRD1-containing aqueous phase is removed so as not to disturb the interphase. To reduce the formation of aggregates, the stock is filtered through sequentially smaller (0.45 micron, 0.22 micron and 0.1 micron), low protein-binding filters pretreated with 5 mL of 0.1% Tween 80 followed by 5 mL DPBS—multiple filters may be necessary to filter the entire PRD1 stock solution. Aliquots of PRD1 bacteriophage stocks are stored at –80°C. 2. Double agar overlay method for PRD1 bacteriophage enumeration: PRD1 bacteriophage are enumerated using the double agar overlay procedure in U.S. EPA Method 1602. In this method, 75μl of prepared bacterial host (i.e. Salmonella typhimurium LT2) and 100μl of PRD1 stock serially diluted in a buffer (DPBS, 0.01% Tween 80 and 0.001% Antifoam-A) are added to a sterile tube containing 5 mL of 0.7% TSA. The contents of the tube are mixed and then poured onto a Petri dish containing 15 mL of 1.5% TSA. The plate, which solidifies within 30 seconds, is then inverted and incubated at 370C for 16-18 hours. During the incubation time, the host bacteria form a confluent lawn over the surface of the Petri plate allowing the phage particles that are present in the sample to bind and penetrate bacterial host cells. The PRD1 bacteriophage then replicates within the host cells causing a subsequent lysis of the host bacterium. The destruction of the bacterial cells that make up the confluent lawn result in clear areas known as plaque forming units. The concentration of bacteriophage present in the sample is determined by visually counting the plaques.
B. Total Coliforms and Escherichia coli
1. E. coli CN-13 stock production: The method for producing E. coli CN-13, a nalidixic acid resistant strain of E. coli, was described previously in the first 12-month reporting period. 2. Enumeration by Colilert®: As stated in the Colilert® Test Kit manual, Colilert simultaneously detects total coliforms and E. coli in water and is based on Defined Substrate Technology® patented by IDEXX. Briefly, contents of one substrate packet is added to a Whirl-Pak® sampling bag containing 100 mL water sample or 0.1% peptone buffer spiked with E. coli CN-13 stock in order to titer the stock. The sampling bag is then shaken until the contents of the packet have dissolved. The sample/reagent mixture is then poured into a Quanti-Tray/2000 and sealed in an IDEXX Quanti-Tray® Sealer to allow for enumeration. The sealed trays are placed at 37° + 0.5°C for 24 hours. The number of yellow wells is recorded, which indicate presence of total coliforms. The trays are placed under a 365nm UV light and the number of wells with fluorescence is recorded—indicative of E. coli. Using the Most Probable Number (MPN) table, a MPN of total coliforms mL sample is determined. This method has been approved by the US EPA for the analysis of wastewater, ambient water, and groundwater for the presence of total coliforms and E. coli (72 FR 14220, 68 FR 43272, and 71 FR 65574, respectively).
C. Enterococcus faecalis (ATCC 29212)
1. Production of E. faecalis stock: E. faecalis is generated by inoculating a culture flask containing 25 mL TSB with a 10-micron loop of frozen E. faecalis stock. The inoculated media is then incubated overnight at 37°C with shaking at 110 rpm. The overnight growth of bacteria is then stored at 4°C for future use. 2. Enumeration of overnight E. faecalis stock
i. Spread Plate: 100μl of bacteria serially diluted in a buffer (same as one used for PRD1) are pipetted onto a Petri dish containing 1% TSA. The diluted sample is spread over the plate by adding four to five 5-mm glass beads to the dish and gently shaking back and forth to allow for even distribution of the bacteria. After allowing the inoculum to absorb into the agar, the plate is then inverted and incubated at 41°C for 16-18 hours. The concentration of E. faecalis present in the sample is determined by visually counting the colony forming units. ii. Enterolert™ MPN: As stated in the Enterolert™ Test Kit manual, Enterolert™ is designed to detect enterococci (E. faecium and E. faecalis) in various types of water and is based on Defined Substrate Technology® patented by IDEXX. Briefly, contents of one substrate packet is added to a Whirl-Pak® sampling bag containing 100 mL water sample or 0.1% peptone buffer spiked with E. faecalis in order to titer the bacterial stock. The sampling bag is then shaken until the contents of the packet have dissolved. The sample/reagent mixture is then poured into a Quanti-Tray/2000 and sealed in an IDEXX Quanti-Tray® Sealer to allow for enumeration. The sealed trays are placed at 41° + 0.5°C for exactly 24 hours. The trays are placed under a 365nm UV light and the number of wells with blue fluorescence is recorded—indicative of enterococci. Using the Most Probable Number (MPN) table, a MPN of enterococci per 100 mL sample is determined. This method has been approved by the US EPA for the analysis of wastewater, ambient water, and groundwater for the presence of enterococci (72 FR 14220, 68 FR 43272, and 71 FR 65574, respectively).
D. Clostridium perfringens
1. Production of C. perfringens stock: To generate C. perfringens spores, 100μl of C. perfringens stock containing approximately 105 colony forming units (cfu) is added to a 50cc polypropylene tube containing 20 mL Duncan-Strong sporulation medium (pH 7.8). The inoculated medium is incubated at 42°C in an anaerobic environment for 48 hours. After incubation, the 50cc tube is centrifuged at 400 x g for 20 minutes in order to pellet bacterial cells. The media is discarded, and the remaining pellet is resuspended in 2 mL DPBS. To differentiate spores from vegetative cells, the new stock is heated at 65°C for 30 minutes. C. perfringens spore stock is then stored at 4°C. 2. Enumeration of C. perfringens stock: C. perfringens stocks are enumerated using a spread plate method. In this method, 100μl of bacteria serially diluted in a buffer (same as one used for PRD1) are pipetted onto a Petri dish containing mCP agar prepared as directed by the manufacturer. The diluted sample is spread over the plate by adding 4 to 5 5-mm glass beads to the dish and gently shaking back and forth to allow for even distribution of the bacteria. After allowing the inoculum to absorb into the agar, the plate is then inverted and incubated at 42°C in an anaerobic environment for 16-18 hours. The concentration of C. perfringens present in the sample is determined by visually counting the colony forming units. For confirmation, the mCP agar plates with yellow colonies—indicative of C. perfringens—are exposed to ammonium hydroxide fumes for 20 seconds. The resulting red and dark pink colonies are counted as presumptive C. perfringens.
Future Activities:
The following research will be conducted during the next project period ( August 2008 - August 2009):
1. Combination of the two previously described multiplexed FISH assays for the detection of G. lamblia, C. parvum, and human-virulent microsporidia will be further evaluated and optimized. Future experiments will include testing incubation temperatures and times to secure optimal binding capacity of combination monoclonal antibodies. In addition, final ultrafiltration confrom spiked water samples will be analyzed for C. parvum oocysts and microsporidian spores using the established FISH assays. This will help to establish to limit of detection for the FISH assay when applied in conjunction with the ultrafiltration method.
2. The next phase of the mass spectrometry work will be to assess the method against well-characterized, real world samples, including final ultrafiltration concentrates, to demonstrate the efficacy and robustness of this method in realistic samples. In addition, we will continue to evaluate and refine the described method to reduce sample preparation complexity and improve the sensitivity of this assay
3. Evaluation of the tangential flow ultrafiltration system will continue. The following parameters will be evaluated:
- The system will be challenged by spiking progressively lower concentrations of microorganisms (i.e. 1,000 CFU and/or PFU total) into 100L samples of dechlorinated tap water and environmental water sample.
- The final ultrafiltration concentrates will continue to be analyzed by both culture and quantitative molecular techniques which have been established in our lab. More specifically, quantitative molecular analysis will be expanded to include all microorganisms used during experimental evaluations.
- As stated previously, additional microorganisms will be added to the water samples including poliovirus, norovirus, Hepatitis A virus, adenovirus, Cryptosporidium and Microsporidia. Our laboratory currently has a culture and/or molecular detection method for each of these microbes. By adding these additional microorganisms, we will be able to further evaluate the ability of the Baxter Exceltra Plus 210 to recovery a wide variety of microorganisms ranging in size, shape, and type.
- 100L surface, ground, and distribution system water samples will be collected and spiked with previously mentioned microorganisms. These samples will then be processed using the ultrafiltration setup and detection methods described within this report.
- 100L environmental water samples will also be processed without the addition of laboratory generated microbes.
- Secondary concentration methods of the final ultrafiltration concentrates will continue to be explored especially for low spike samples and environmental water samples. Current methods under investigation include 1) the use of 100kDa MWCO centrifugal filtration devices and 2) polyethylene glycol concentration followed by TRIzol extraction and ethanol precipitation of nucleic acids.
References:
1. Polaczyk, A., Narayanan, J., Cromeans, T., Hahn, D., Roberts, J., Amburgey, J., & Hill, V. Ultrafiltration-based techniques for rapid and simultaneous concentration of multiple microbe classes from 100-L tap water samples. Journal of Microbiological Methods 73, 92-99 (2008)
2. Heid, C., Stevens, J., Livak, K. & Williams, P. Real time quantitative PCR. Genome Research 6, 986-994 (1996).
Journal Articles on this Report : 4 Displayed | Download in RIS Format
| Other project views: | All 8 publications | 4 publications in selected types | All 4 journal articles |
|---|
| Type | Citation | ||
|---|---|---|---|
|
|
Graczyk TK, Sunderland D, Rule AM, da Silva AJ, Moura INS, Tamang L, Girouard AS, Schwab KJ, Breysse PN. Urban feral pigeons (Columba livia) as a source for air- and waterborne contamination with Enterocytozoon bieneusi spores. Applied and Environmental Microbiology 2007;73(13):4357-4358. |
R833002 (2007) R833002 (2008) R833002 (Final) |
Exit Exit Exit |
|
|
Graczyk TK, Majewska AC, Schwab KJ. The role of birds in dissemination of human waterborne enteropathogens. Trends in Parasitology 2008;24(2):55-59. |
R833002 (2007) R833002 (2008) R833002 (Final) |
Exit Exit |
|
|
Graczyk T K, Conn DB. Molecular markers and sentinel organisms for environmental monitoring. Parasite 2008;15(3):458-462. |
R833002 (2008) |
Exit |
|
|
Young TA, Heidler J, Matos-Perez CR, Sapkota A, Toler T, Gibson KE, Schwab KJ, Halden RU. Ab initio and in situ comparison of caffeine, triclosan, and triclocarbon as indicators of sewage-derived microbes in surface waters. Environmental Science & Technology 2008;42(9):3335-3340. |
R833002 (2008) R833002 (Final) |
Exit Exit |
Supplemental Keywords:
drinking water, human health, molecular detection, monitoring, quantitative PCR, FISH, proteomics, mass spectrometry, pathogens, viruses, protozoa, bacteria, exposure, risk assessment, environmental microbiology, Maryland (MD), California (CA), POLLUTANTS/TOXICS, Water, INTERNATIONAL COOPERATION, Scientific Discipline, RFA, PHYSICAL ASPECTS, Drinking Water, Physical Processes, Environmental Engineering, Environmental Chemistry, Environmental Monitoring, Microorganisms, bacteria, drinking water contaminants, mass spectrometry, pathogenic protozoa, molecular detection, viruses, pathogens, ultrafiltration, exposure, human health,, RFA, Scientific Discipline, PHYSICAL ASPECTS, INTERNATIONAL COOPERATION, Water, POLLUTANTS/TOXICS, Physical Processes, Environmental Chemistry, Microorganisms, Drinking Water, Environmental Engineering, Environmental Monitoring, human health, water quality, viruses, mass spectrometry, ultrafiltration, pathogens, pathogenic protozoa, drinking water contaminants, exposure, drinking water monitoring, molecular detectionProgress and Final Reports:
Original AbstractThe perspectives, information and conclusions conveyed in research project abstracts, progress reports, final reports, journal abstracts and journal publications convey the viewpoints of the principal investigator and may not represent the views and policies of ORD and EPA. Conclusions drawn by the principal investigators have not been reviewed by the Agency.