
Grantee Research Project Results
Final Report: Diesel-induced Vascular Dysfunction: Role of Endothelin
EPA Grant Number: R831860Title: Diesel-induced Vascular Dysfunction: Role of Endothelin
Investigators: Kanagy, Nancy L. , Campen, Matthew J. , Walker, Benjimen R.
Institution: University of New Mexico , Lovelace Biomedical & Environmental Research Institute
EPA Project Officer: Chung, Serena
Project Period: October 1, 2004 through September 30, 2008 (Extended to September 30, 2010)
Project Amount: $1,500,000
RFA: The Role of Air Pollutants in Cardiovascular Disease (2003) RFA Text | Recipients Lists
Research Category: Particulate Matter , Air Quality and Air Toxics , Human Health , Air
Objective:
Recent studies have substantiated the premise of this proposal that air pollution exposure is associated with cardiovascular morbidity and mortality. In addition, a number of reports have demonstrated that a portion of this is due to alterations in vascular and cardiac function. Multiple inhaled pollutants have been shown to decrease the ability of vascular endothelium to release vasodilator substances and to increase the production of vasoactive cytokines such as endothelin. Therefore we have been examining specific cellular changes associated with exposure to diesel exhaust. We proposed to use a novel model of endothelin-dependent hypertension and endothelial dysfunction, paired with state-of-the-art methods for generating whole diesel exhaust, to investigate cardiovascular effects of PM.
These studies were designed to determine the mechanisms for intermittent hypoxia (a rat model of sleep apnea, IH) and air pollution to synergistically augment ET-dependent vasoconstriction with a focus on the generation of reactive oxygen species (ROS). ROS are a potent stimulus for ET synthesis and augment ET vasoconstriction. Others have demonstrated ROS generation by multiple pollutants and our preliminary data show that diesel exposure alone stimulates the generation of ROS. Therefore, DE exposure may increase ROS-dependent release of ET and further promote ET-dependent vasoconstriction in already compromised individuals. Our central hypothesis was that inhalation of whole DE augments ET-vasoconstriction in ET-sensitized hypertension. Studies carried out under each of the original aims are described below.
Objective 1: Identify the role of ET in DE-induced vasoconstriction in IH and Sham rats.
Hypothesis:Inhalation of whole DE releases ET to increase vascular resistance, blood pressure, and venous return in IH rats but not in Sham rats.
The initial studies established the time course of increases in plasma and tissue ET and ET-receptors following acute inhalation of whole DE and in chronic IH. Those studies were completed in the first year of the grant and demonstrated that there was only a modest increase in ET-1 synthesis and only in a few tissues for DE and that there was no apparent synergy between the two conditions. This was in contrast to earlier studies using much higher concentrations of DE. However, parallel studies in mice found that there is a cumulative increase in ET-1 synthesis (8) validating the focus on this peptide. Additional studies evaluated the changes in hemodynamics and found evidence of sympathetic activation following acute exposure to DE but no significant changes in arterial pressure and no interaction with IH. The data were largely negative (i.e. no large changes and no interactions) and no additional studies are being conducted under this aim but the data from these studies are part of a manuscript that was published describing the effects of IH alone(1) and the sympathetic nervous system activation by DE data are being re-written for re-submission for publication.
Objective 2: Identify the effect of inhaled whole DE on vascular smooth muscle and endothelial cell function in rats.
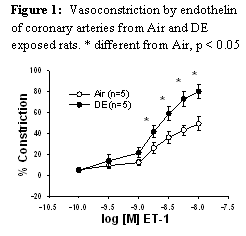
Hypothesis: DE inhalation augments ET vasoconstriction and decreases endothelium-induced vasodilation in IH rats but not in Sham rats.
Studies under aim 2 were the primary focus of research in the study period. Vasoconstrictor responses to ET were evaluated in air and DE exposed rats to determine the mechanism for the increased constrictor response to this peptide (Figure 1). We found that removal of the endothelium, inhibition on nitric oxide synthase or scavenging of reactive oxygen normalized the response. We also observed that adding NOS co-factor, tetrahydorbiopterin, normalized the response. We therefore concluded that DE exposure caused increased coronary artery reactivity by increasing reactive oxygen inhibition of endothelial function. These studies were published in the American Journal of Physiology in July 2009(3).
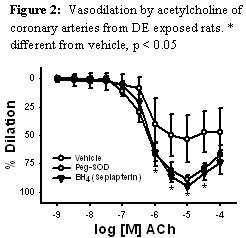
A second set of studies was completed in this past year investigating the cellular mechanism of this dysfunction. We observed that DE impairment of nitric oxide production limited dilation in the coronary arteries and that this could be improved by scavenging the reactive oxygen with a cell permeable antioxidant, PEG-SOD, or by supplementing the critical co-factor for nitric oxide production, tetrahydrobiopterin (BH4. Figure 2). These studies were published in Environmental Health Perspectives in 2010. (4)
Summary/Accomplishments (Outputs/Outcomes):
The lack of a clear synergism between the two conditions (DE exposure and IH) led us to focus on the effects of diesel alone in control rats. It also suggested that sleep apnea may not predispose individuals to DE-induced cardiac pathology. We did observed that following exposure to DE, ET-1 constriction was greatly augmented and endothelium-dependent dilation was profoundly decreased in healthy rats suggesting that this is a general mechanism.
Studies focusing on the mechanism for that change determined that impaired coronary artery function after DE inhalation appears to be ROS-dependent uncoupling of eNOS. This was verified by using inhibitors of NOS and measures of ROS in isolated arteries.
To address the role of NOS uncoupling in the augmented constriction to eT-1, we gave excess NOS substrate, BH4 in the form of sepiapterin and also scavenged the generated ROS using cell permeable antioxidant, PEG-SOD. Both of these interventions restored normal function in the DE exposed arteries suggesting that this is indeed the mechanism of the impaired function (figure 2). These studies were presented at two conferences, a Gordon conference and the Society of Toxicology Meetings where Mr. Cherng received awards for the presentations (see below) and published in Environmental Health Perspectives in 2010 (4). In addition, studies from this proposal were published in several reviews (5-7)
References:
- Allahdadi KJ, Cherng TW, Pai H, Silva AQ, Walker BR, Nelin LD and Kanagy NL. Endothelin type A receptor antagonist normalizes blood pressure in rats exposed to eucapnic intermittent hypoxia. Am J Physiol Heart Circ Physiol 295: H434-H440, 2008.
- Bosc LV, Resta T, Walker B and Kanagy NL. Mechanisms of intermittent hypoxia induced hypertension. J Cell Mol Med 14: 3-17, 2010.
- Cherng TW, Campen MJ, Knuckles TL, Gonzalez Bosc LV and Kanagy NL. Impairment of coronary endothelial cell ETB receptor function following short-term inhalation exposure to whole diesel emissions. Am J Physiol Regul Integr Comp Physiol 2009.
- Cherng TW, Paffett ML, Jackson-Weaver O, Campen MJ, Walker BR and Kanagy NL. Mechanisms of Diesel-Induced Endothelial Nitric Oxide Synthase Dysfunction in Coronary Arterioles. Environ Health Perspect 2010.
- da Silva AQ, Fontes MA and Kanagy NL. Chronic infusion of angiotensin receptor antagonists in the hypothalamic paraventricular nucleus prevents hypertension in a rat model of sleep apnea. Brain Res 2010.
- Kanagy NL. Vascular effects of intermittent hypoxia. ILAR J 50: 282-288, 2009.
- Kanagy NL, Pawloski CM and Fink GD. Role of aldosterone in angiotensin II-induced hypertension in rats. Am J Physiol 259: R102-R109, 1990.
- Lund AK, Knuckles TL, Obot AC, Shohet R, McDonald JD, Gigliotti A, Seagrave JC and Campen MJ. Gasoline exhaust emissions induce vascular remodeling pathways involved in atherosclerosis. Toxicol Sci 95: 485-494, 2007.
Journal Articles on this Report : 14 Displayed | Download in RIS Format
| Other project views: | All 22 publications | 14 publications in selected types | All 14 journal articles |
|---|
| Type | Citation | ||
|---|---|---|---|
|
|
Allahdadi KJ, Walker BR, Kanagy NL. Augmented endothelin vasoconstriction in intermittent hypoxia-induced hypertension. Hypertension 2005;45(4):705-709. |
R831860 (2008) R831860 (Final) |
Exit Exit Exit |
|
|
Allahdadi KJ, Walker BR, Kanagy NL. ROK contribution to endothelin-mediated contraction in aorta and mesenteric arteries following intermittent hypoxia/hypercapnia in rats. American Journal of Physiology-Heart and Circulatory Physiology 2007;293(5):H2911-H2918. |
R831860 (2008) R831860 (Final) |
Exit Exit Exit |
|
|
Allahdadi KJ, Duling LC, Walker BR, Kanagy NL. Eucapnic intermittent hypoxia augments endothelin-1 vasoconstriction in rats: role of PKCδ. American Journal of Physiology-Heart and Circulatory Physiology 2008;294(2):H920-H927. |
R831860 (2008) R831860 (Final) |
Exit Exit Exit |
|
|
Allahdadi KJ, Cherng TW, Pai H, Silva AQ, Walker BR, Nelin LD, Kanagy NL. Endothelin type A receptor antagonist normalizes blood pressure in rats exposed to eucapnic intermittent hypoxia. American Journal of Physiology-Heart and Circulatory Physiology 2008;295(1):H434-H440. |
R831860 (2008) R831860 (Final) |
Exit Exit Exit |
|
|
Bosc LVG, Resta T, Walker B and Kanagy NL. Mechanisms of intermittent hypoxia induced hypertension. Journal of Cellular and Molecular Medicine 2010;14(1-2):3-17, 2010. |
R831860 (Final) |
Exit Exit |
|
|
Cherng TW, Campen MJ, Knuckles TL, Gonzalez Bosc L, Kanagy NL. Impairment of coronary endothelial cell ETB receptor function after short-term inhalation exposure to whole diesel emissions. American Journal of Physiology. Regulatory, Integrative, and Comparative Physiology 2009;297(3):R640-R647. |
R831860 (2008) R831860 (Final) |
Exit Exit Exit |
|
|
Cherng TW, Paffett ML, Jackson-Weaver O, Campen MJ, Walker BR, Kanagy NL. Mechanisms of diesel-induced endothelial nitric oxide synthase dysfunction in coronary arterioles. Environmental Health Perspectives 2011;119(1):98-103. |
R831860 (Final) |
|
|
|
Duling LC, Cherng TW, Griego JR, Perrine MF, Kanagy NL. Loss of α2B-adrenoceptors increases magnitude of hypertension following nitric oxide synthase inhibition. American Journal of Physiology-Heart and Circulatory Physiology 2006;291(5):H2403-H2408. |
R831860 (2008) R831860 (Final) |
Exit Exit Exit |
|
|
Gomes da Silva AQ, Fontes MA, Kanagy NL. Chronic infusion of angiotensin receptor antagonists in the hypothalamic paraventricular nucleus prevents hypertension in a rat model of sleep apnea. Brain Research 2010;1368:231-238. |
R831860 (Final) |
Exit Exit Exit |
|
|
Kanagy NL. α2-adrenergic receptor signalling in hypertension. Clinical Science (London) 2005;109(5):431-437. |
R831860 (2008) R831860 (Final) |
Exit Exit |
|
|
Kanagy NL. Vascular effects of intermittent hypoxia. ILAR Journal 2009;50(3):282-288. |
R831860 (Final) |
|
|
|
Lund AK, Knuckles TL, Akata CO, Shohet R, McDonald JD, Gigliotti A, Seagrave JC, Campen MJ. Gasoline exhaust emissions induce vascular remodeling pathways involved in atherosclerosis. Toxicological Sciences 2007;95(2):485-494. |
R831860 (2007) R831860 (Final) CR831455 (Final) R830839 (2005) R830839 (Final) |
Exit Exit Exit |
|
|
Snow JB, Kitzis V, Norton CE, Torres SN, Johnson KD, Kanagy NL, Walker BR, Resta TC. Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. Journal of Applied Physiology 2008;104(1):110-118. |
R831860 (2008) R831860 (Final) |
Exit Exit Exit |
|
|
Troncoso Brindeiro CM, da Silva AQ, Allahdadi KJ, Youngblood V, Kanagy NL. Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. American Journal of Physiology-Heart and Circulatory Physiology 2007;293(5):H2971-H2976. |
R831860 (2008) R831860 (Final) |
Exit Exit Exit |
Supplemental Keywords:
Sleep apnea, diesel exhaust, toxicology, endothelin, blood pressure, cardiac, coronary, endothelial dysfunction, autonomic nervous system, sympathetic nervous system, heart rate variability, reactive oxygen species, superoxide, hydrogen peroxide, SOD, catalase;, RFA, Health, Scientific Discipline, Air, PHYSICAL ASPECTS, HUMAN HEALTH, Susceptibility/Sensitive Population/Genetic Susceptibility, Ecological Risk Assessment, Health Risk Assessment, Physical Processes, Risk Assessments, particulate matter, Biology, genetic susceptability, mobile sources, Environmental Chemistry, Exposure, Toxicology, air toxics, air quality, diesel exhaust, inhaled, sensitive populations, environmental hazard exposures, inhaled pollutants, fine particles, lung inflammation, oxidant gas, engine exhaust, acute lung injury, endothelial function, DEP, cardiopulmonary responses, particulate exposure, acute exposure, Acute health effects, cardiotoxicity, copollutant exposures, airway epithelial cells, diesel exhaust particulate, chronic health effects, susceptible subpopulations, toxics, concentrated particulate matter, heart rate, air contaminant exposure, atmospheric particulate matter, air pollution, cardiopulmonary, cardiovascular disease, highrisk groups, human susceptibility, airborne urban contaminants, ambient particle pollution, automotive exhaust, cardiopulmonary response, diesel exhaust particlesProgress and Final Reports:
Original AbstractThe perspectives, information and conclusions conveyed in research project abstracts, progress reports, final reports, journal abstracts and journal publications convey the viewpoints of the principal investigator and may not represent the views and policies of ORD and EPA. Conclusions drawn by the principal investigators have not been reviewed by the Agency.