
Grantee Research Project Results
Final Report: Sensitivity of Organic Aerosol Concentrations and Forcing to Anthropogenic Emissions
EPA Grant Number: R835405Title: Sensitivity of Organic Aerosol Concentrations and Forcing to Anthropogenic Emissions
Investigators: Pandis, Spyros N. , Donahue, Neil
Institution: Carnegie Mellon University
EPA Project Officer: Chung, Serena
Project Period: April 1, 2013 through March 31, 2016
Project Amount: $399,998
RFA: Anthropogenic Influences on Organic Aerosol Formation and Regional Climate Implications (2012) RFA Text | Recipients Lists
Research Category: Air Quality and Air Toxics , Air , Climate Change
Objective:
Summary/Accomplishments (Outputs/Outcomes):
Organic Aerosol Mixing: The partitioning of organic pollutants between the gas and articulate phases plays a critical role in determining the chemical composition and physical characteristics of atmospheric particles. Most regional-scale chemical transport models (CMAQ, CAMx, etc.) assume an internally mixed aerosol. This assumption means that all particles of the same size have the same chemical composition. In this project, we have developed novel experimental techniques for performing aerosol mixing experiments using single particle mass spectrometry for determining the mixing of progressively more complex organic aerosol systems. The various stages of the mixing experiments are shown in Figure 1.
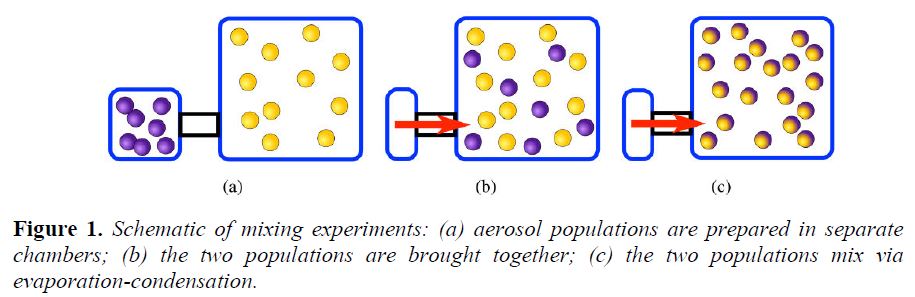
Single-particle mass spectra from the high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) are collected and their degree of similarity with time was examined. Depending on the similarity the mixing behavior and timescales can be determined (Figure 2).
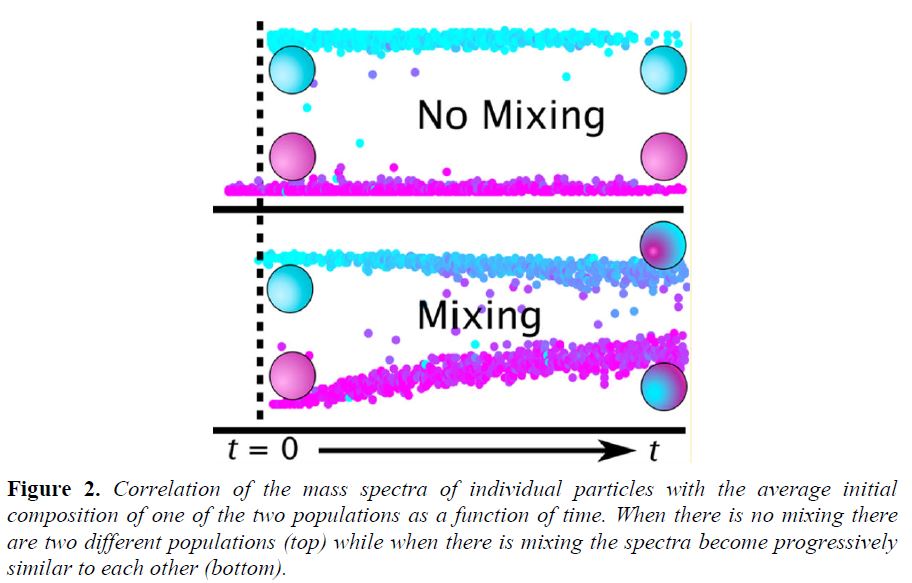
Docosane and docosane-d46 (22 carbon linear solid alkane) did not show any signs of mixing, but squalane and squalane-d62 (30 carbon branched liquid alkane) mixed on the time scale expected from a condensational-mixing model. Docosane and docosane-d46 were driven to mix when the chamber temperature was elevated above the melting point for docosane. Docosane vapors were shown to mix into squalane-d62, but not the other way around. These results are consistent with low diffusivity in the solid phase of docosane particles.
We performed mixing experiments on secondary organic aerosol (SOA) surrogate systems finding that SOA derived from toluene-d8 (a surrogate for anthropogenic SOA (aSOA)) does not mix into squalane (a surrogate for hydrophobic primary organic aerosol (POA)) but does mix into SOA derived from α-pinene (biogenic SOA (bSOA) surrogate). For the aSOA/POA, the volatility of either aerosol does not limit gas-phase diffusion, indicating that the two particle populations do not mix simply because they are immiscible. In the aSOA/bSOA system, the presence of toluene-d8-derived SOA molecules in the α-pinene-derived SOA provides evidence that the diffusion coefficient in α-pinene-derived SOA is high enough for mixing on the time scale of 1 min.
The formation and evaporation of mixed particles containing squalane (a surrogate for hydrophobic primary organic aerosol, POA) and secondary organic aerosol (SOA) was also examined. In these experiments, one material (D62-squalane or SOA from α-pinene + O3) was prepared first to serve as surface area for condensation of the other, forming the mixed-particles. The mixed-particles were then subjected to a heating ramp from 22 to 44 °C. We were able to determine that (1) almost all of the SOA mass is comprised of material less volatile than D62-squalane; (2) AMS collection efficiency in these mixed-particle systems can be parameterized as a function of the relative mass fraction of the components; and (3) the vast majority of D62-squalane is able to evaporate from the mixed particles, and does so on the same time scale regardless of the order of preparation.
We also performed two-population mixing experiments to directly test whether D62-squalane and SOA from α-pinene + O3 form a single solution or two separate phases. We find that these two OA types are immiscible, which informs our inference of the morphology of the mixed-particles. If the morphology is core−shell and dictated by the order of preparation, these data indicate that squalane is able to diffuse relatively quickly through the SOA shell, implying that there are no major diffusion limitations.
Oxidation of organic compounds, carried to completion, will form CO2, and consequently oxidation is fundamentally destructive to organic aerosol. However, oxidation clearly is a source of organic aerosol as well. Consequently, organic aerosols can only be viewed as metastable intermediates. Its concentrations are thus controlled by its lifetime, and we have been able to show the fundamental governor of this lifetime is the delay caused by diffusion of OH radicals to aerosols. Without this, organic aerosol levels would be many times lower than they are, rendering organics virtually irrelevant to aerosol concentrations and properties. Conversely, although organic compounds can run, they cannot hide. Sequestration into the condensed phase delays oxidation because of diffusion limitations in the gas phase, but heterogeneous oxidation is still important. The near complete oxidation observed in situations where transport occurs within 1 day from intense sources indicates that the diffusion timescale for organics within particles is usually less than 1 day. These results have been presented in Donahue, et al. [2013].
Chemical Aging of Organic Aerosol: Monoterpenes are a significant class of volatile organic compounds emitted by vegetation and β-caryophyllene is one of the most important compounds in this class. The secondary organic aerosol (SOA) production during the oxidation of β-caryophyllene by ozone (O3) and hydroxyl radicals (OH) and the subsequent chemical aging of the products during reactions with OH were investigated. Experiments were conducted with ozone, hydroxyl radicals at low NOx and at high NOx (100s of ppb). The SOA mass yield at 10 μg m-3 of organic aerosol was 27% for the ozonolysis, 20% for the reaction with OH at low NOx and 38% at high NOx under dry conditions, 20o C, and ozone excess. Parameterizations of the fresh SOA yields have been developed. The average fresh SOA atomic O:C ratio varied from 0.24 to 0.34 depending on the oxidant and the NOx level, while the H:C ratio was close to 1.5 for all systems examined. An average density of 1.06 ± 0.1 μg m-3 of the β-caryophyllene SOA was estimated. The exposure to UV-light had no effect on the β-caryophyllene SOA concentration and Aerosol Mass Spectrometer (AMS) mass spectrum. The chemical aging of the produced β- caryophyllene SOA was studied by exposing the fresh SOA to high concentrations (107 molecules cm-3) of OH for several hours. These additional reactions increased the SOA concentration by 15-40% and the O:C by approximately 25%. A limited number of experiments suggested that there was a significant impact of the relative humidity on the chemical aging of the SOA. The evaporation rates of β-caryophyllene SOA were quantified by using a thermodenuder allowing us to estimate the corresponding volatility distributions and effective vaporization enthalpies. Details about this work can be found in Tasoglou, et al. [2015].
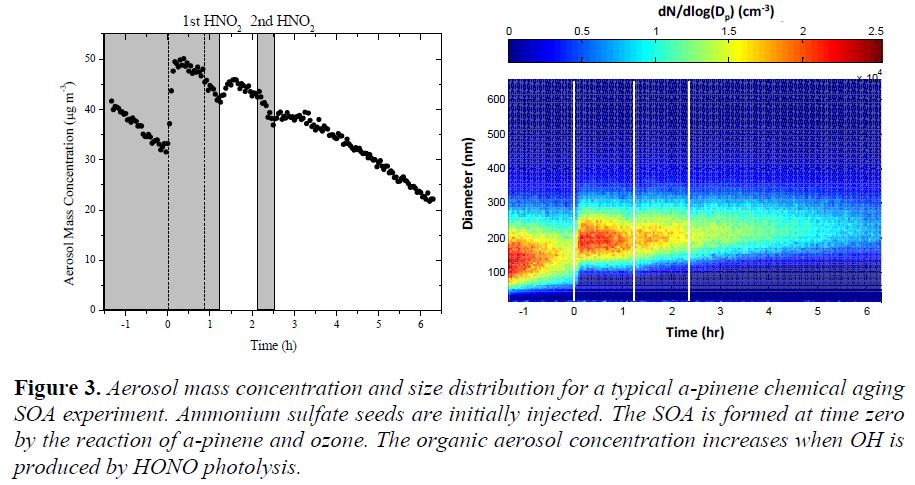
Secondary organic aerosol (SOA) formation from volatile organic compounds in the atmosphere can be thought of as a succession of oxidation steps. The production of later generation SOA via continued oxidation of the first-generation products is defined as chemical aging. The chemical aging of SOA produced during the α-pinene ozonolysis was investigated in a series of smog chamber experiments. After the SOA production, the resulting particles and vapors were exposed to OH radicals at both low and high NOx levels. In the former experiments H2O2 photolysis was the OH source, while in the latter HONO was photolyzed producing OH. Up to approximately 30% increase in organic aerosol (OA) mass was observed after an equivalent of 10 h of ambient daytime exposure to OH. A more oxygenated product distribution was also observed after aging based on the increase in aerosol atomic oxygen to carbon ratio. Experiments exposing the first-generation α-pinene ozonolysis products to UV lights only showed a small reduction in OA mass, indicating minimum photolysis effect for this system. Experiments performed at elevated relative humidity (RH) of 55% showed no significant difference in additional SOA formation after the aging step compared to those performed at low RH of ≤ 20%. Experiments with OH introduction at different times after the first-generation SOA formation yielded similar additional SOA mass, indicating direct vapor loss to the chamber walls is minimal under these circumstances. Experiments at high NOx conditions resulted in general in higher SOA production during the chemical aging steps. Results of a typical experiment are shown in Figure 3. An increase in organic aerosol concentration was observed after the two HONO injections when the lights were turned on. A parameterization for the description of the aging effects on aerosol yield was developed. The SOA concentration increases were however modest. This work is described in Wang, et al., in press.
The chemical aging of anthropogenic SOA was also investigated focusing mainly on toluene as a precursor. Photochemical aging clearly influences anthropogenic SOA, and the general trend toward increased SOA mass and reduced volatility is consistent with progressive oxidation driving organic aerosol toward the highly oxidized, low-volatility endpoint observed around the world. There is a strong relationship between exposure to OH and physicochemical properties for SOA formed from the oxidation of toluene and other small aromatic VOCs. Organic nitrogen compounds were a major constituent in the SOA formed. An experiment with higher OH exposure showed higher SOA mass yields, more oxidized SOA, and reduced SOA volatility but only modest differences in hygroscopicity. Volatility varied by a factor of 30 for different OH exposure, and a ten-fold decrease in volatility was associated with a 0.3 increase in carbon oxidation state. The SOA was relatively hygroscopic for organic material, with 0.1 < κ < 0.2 and if anything a slightly negative relationship between kappa and oxidation state, suggesting a possible role for surfactants or oligomeric compounds. While individual experiments with different OH exposure showed clear aging effects as different oxidation states and OA volatility, these effects were not evident within every single experiment. This suggests that a complex interplay exists between gas-phase processes, including oxidation reactions that both functionalize and fragment condensable organic species as well as photolysis of some species. The composition, hygroscopicity and volatility of organic aerosol do not follow a prescribed relationship, and additional studies are needed which evaluated all of these properties in future laboratory experiments and ambient measurements. This work has been published as Hildebrandt-Ruiz, et al. [2015].
Field Perturbation Experiments: We have developed a mobile dual smog chamber system for field perturbation experiments. The system is shown in Figure 4. It can be set-up in less than 2 hours outdoors and experiments can be performed using ambient sunlight, during the night using artificial light, or in the dark during day or night. In these experiments the starting point is ambient air. Both chambers are filled with ambient air and after its chemical characterization a perturbation is imposed in the first chamber. The second is used as the baseline. Instruments housed in a mobile laboratory sample alternatively both chamber.
In the test experiment the experimental system was deployed in a park. Characterization experiments included blank experiments, measurements of losses of particles, testing of the similarity of the results in the two chambers using only ambient air (no perturbation), etc. The air was characterized with a combination of aerosol (SMPS, APS, AMS) and gas-phase instruments (PTR-MS, O3, NOx, SO2, etc., monitors).
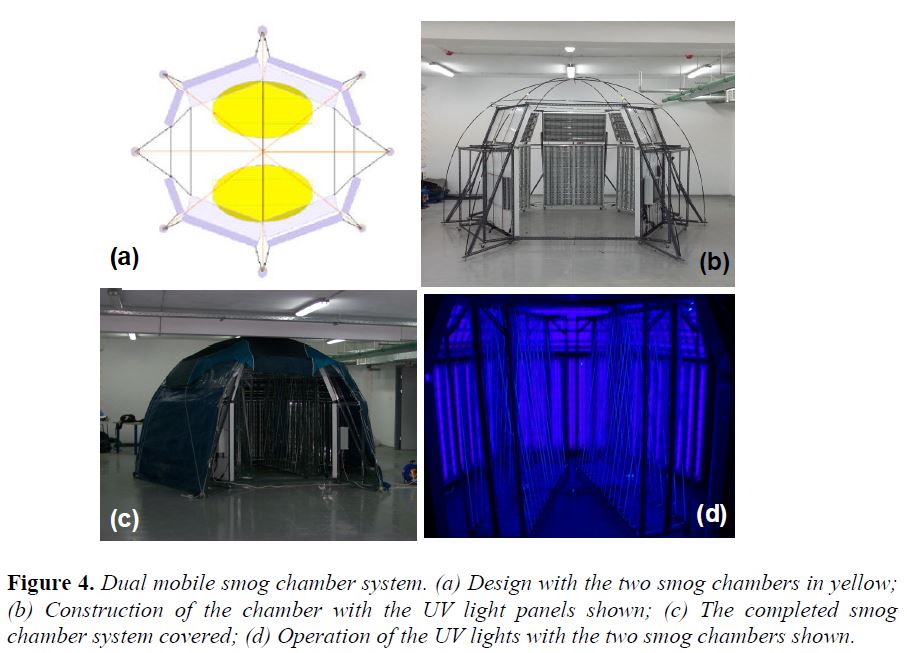
In a pilot study α-pinene was added to the first chamber to investigate its SOA formation in a mixture with ambient urban air. The measured organic aerosol concentration is shown in Figure 5. At 16:00 ambient air existed in both chambers. A few ppb of α-pinene was added in Chamber 1 at that point and it was allowed to react with the existing ozone in the dark. The reaction resulted in the formation of a little more than 1 μg m-3 of SOA in Chamber 1 (after the wall loss corrections) while, as expected there were negligible changes in Chamber 2. The calculated SOA for this experiment were consistent with laboratory measurements using only α-pinene in clean air.
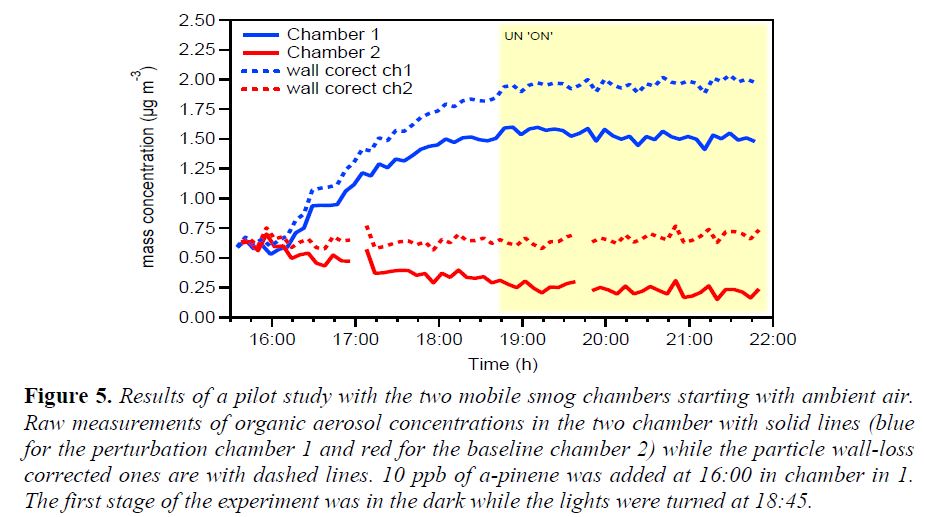
The system development and evaluation are presented in Kaltsonoudis, et al., in press.
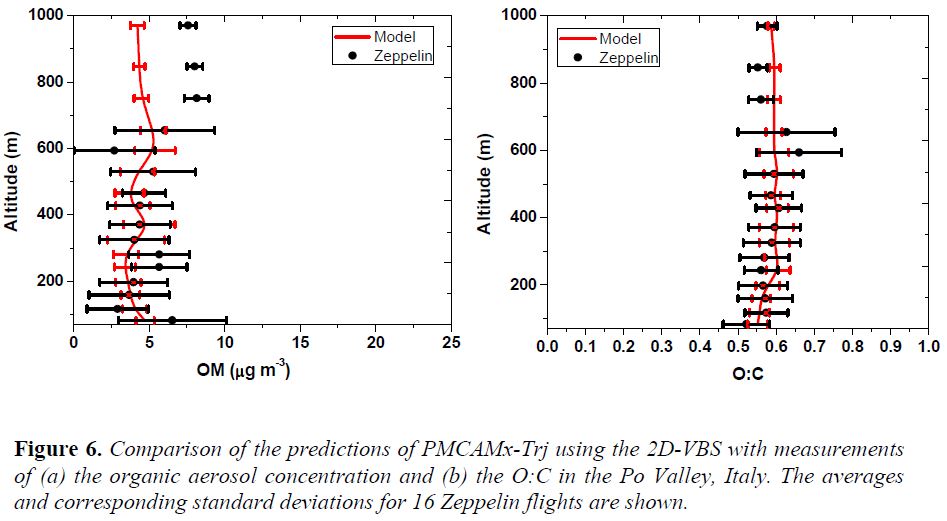
OA Module Development and Evaluation: The organic aerosol modules in PMCAMx and PMCAMx-Trj (the 1-D version of the CTM using the 2D-VBS) have been updated using the yields and chemical aging parameterizations developed in the first year of the project. Additional updates are expected based on the third year of the experiments. We have continued the evaluation and uncertainty analysis of these CTMs using the reach dataset collected during the PEGASOS-2012 campaign in Central and Southern Europe. One special feature of the dataset is the use of a Zeppelin for the collection of measurements of pollutant concentrations aloft.
The model evaluation suggests that using the oxygen to carbon (O:C) ratio together with the organic aerosol concentration provides additional constraints to the various organic aerosol schemes that are currently used in CTMs. The original Volatility Basis Set scheme with its simplistic treatment of SOA production and aging (it simulates only the average net effect of functionalization and fragmentation) performed well in both reproducing the OA levels but also the O:C ratio (Figure 6) both at the ground and aloft. A number of alternative formulations of the 2D-VBS were tested and a number of them could reproduce relatively well the same observations while others failed especially to reproduce the O:C observations. The schemes that reproduced the observations gave similar results about the importance of anthropogenic SOA from VOCs (22 ± 7 % of the total OA), fresh POA (around 10%), SOA from the oxidation of anthropogenic semivolatile compounds (around 10%), but differed on the relative importance of biogenic SOA (from a low of 20 to a high of 50% of the total OA depending on the parameters) and SOA from anthropogenic IVOCs (from 20 to 35%).
This work is described in Karnezi and Pandis [2016] currently in preparation.
Organic aerosol emissions, formation and removal: Five case studies (Pittsburgh and Los Angeles in the United States, Athens and Paris in Europe, and Mexico City in Central America) were used to gain insights about the changing levels, sources, and role of atmospheric chemical processes on air quality in large urban areas as they develop technologically. Fine particulate matter was the focus of our analysis. In all cases reductions of emissions by industrial and transportation sources have resulted in significant improvements in air quality during the last decades. However, these changes have resulted in increasing importance of secondary particulate matter (PM) that dominates over primary in most cases. At the same time long range transport of secondary PM from sources located hundreds of kilometers from the city center is becoming a bigger contributor to the urban PM levels in all seasons. “Non-traditional” sources including cooking, residential and agricultural biomass burning contribute an increasing fraction of the now reduced fine PM levels. Atmospheric chemistry is found to change the chemical signatures of a number of these sources relatively fast both during day and night complicating the corresponding source apportionment [Pandis, et al., 2016].
We have proposed a naming system for classifying atmospheric organic particle and gas compounds. This system is consistent with general classifications used in many field and laboratory studies, as well as developing model frameworks. Specifically, it is applicable to the current conceptual model of quite dynamic mechanisms by which these particles form and age. By introducing a standardized rule for communicating phase state via subscript, the scheme emphasizes the importance of phase transitions and dynamic interactions observed by the scientific community while maintaining consistency with the terms used to communicate to the policy community. We have chosen a classification system that segregates compounds based on their effective saturation concentration at 298 K. The use of alphabetic prefixes (e.g., ELV-, LV-, SV-, etc.) is standardized in terms of the effective saturation concentration C*. We have also added a lowercase suffix to track the volatility of each species when it was emitted. This suffix can be applied to any root term describing the chemical nature of OA (e.g., POA, BBOA, aSOA, etc.) and bridges the gap between the traditional, static view of the POA/SOA system and the more recent, dynamic view which treats evaporation upon dilution and aging of both primary and secondary material. Because of the observed semivolatile behavior of POA from many emissions sources, it is important to standardize the conditions at which particles will be defined to be primary. We propose this to be at 298 K and an OA concentration of 320 μg m−3. This concentration is a suitable compromise between the low loadings seen at ambient conditions and the higher loadings often encountered when performing source measurements. It also agrees nicely with the proposed division between semivolatile and intermediate volatility OA. This framework provides a standard for communicating detailed phase, volatility, source, and chemical information to experts and non-experts alike, and will be useful as the field continues to evolve. This framework is described in Murphy, et al. [2014].
The elemental carbon (EC) tracer method has often been used to estimate the primary and secondary organic aerosol (OA) fractions using field measurements of organic carbon (OC) and EC. In this observation-based approach, EC is used as a tracer for primary OC (POC), which allows for the estimation of secondary OC (SOC). The accuracy of this approach was evaluated using concentrations generated by PMCAMx, a three-dimensional chemical transport model that simulates the complex processes leading to SOC formation (including evaporation and chemical processing of POC and chemical aging of semivolatile and intermediate volatility organics). The ratio of primary organic to elemental carbon [OC/EC]p was estimated in various locations in the Eastern United States, and was then used to calculate the primary and secondary OC concentrations. To estimate the [OC/EC]p from simulated concentrations, we used both a traditional approach and the high EC edge method, in which only values with the highest EC/OC ratio are used. Both methods performed best on a daily-averaged basis, because of the variability of the [OC/EC]p ratio during the day. The SOC estimated by the EC tracer methods corresponds to the biogenic and anthropogenic SOC formed during the oxidation of volatile organic compounds. On the other hand, the estimated POC corresponds to the sum of the fresh POC, the SOC from oxidation of the evaporated POC and the intermediate volatility organic compounds, and the OC from long-distance transport. With this correspondence, the traditional EC tracer method tends to overpredict primary OC and underpredict secondary OC for the selected urban areas in the eastern United States. The high EC edge method performs better, especially in areas where the primary contribution to OC is smaller. Details can be found in Day et al. [2015].
Organics, particle number and aerosol optical properties: Source contributions to ultrafine particle number concentrations for a summertime period in the Eastern U.S. are investigated using the chemical transport model PMCAMx-UF. New source-resolved number emissions inventories were developed for biomass burning, dust, gasoline automobiles, industrial sources, non-road and on-road diesel. According to the inventory for this summertime period in the Eastern U.S., gasoline automobiles are responsible for 40% of the ultrafine particle number emissions, followed by industrial sources (33%), nonroad diesel (16%), on-road diesel (10%), and 1% from biomass burning and dust. With these emissions as input, the chemical transport model PMCAMx-UF reproduces observed ultrafine particle number concentrations (N3-100) in Pittsburgh with an error of 12%. For this summertime period in the Eastern U.S., nucleation is predicted to be the source of more than 90% of the total particle number concentrations. The source contributions to primary particle number concentrations are on average similar to those of their source emissions contributions: gasoline is predicted to contribute 36% of the total particle number concentrations, followed by industrial sources (31%), non-road diesel (18%), on-road diesel (10%), biomass burning (1%), and long-range transport (4%). For this summertime period in Pittsburgh, number source apportionment predictions for particles larger than 3 nm in diameter (traffic 65%, other combustion sources 35%) are consistent with measurement-based source apportionment (traffic 60%, combustion sources 40%). This work was published as Posner, et al. [2015].
An experimental methodology was developed to measure the nonvolatile particle number concentration using a thermodenuder (TD). The TD was coupled with a high resolution time-of-flight aerosol mass spectrometer, measuring the chemical composition and mass size distribution of the submicrometer aerosol and a scanning mobility particle sizer (SMPS) that provided the number size distribution of the aerosol in the range from 10 to 500 nm. The method was evaluated with a set of smog chamber experiments and achieved almost complete evaporation (> 98 %) of secondary organic as well as freshly nucleated particles, using a TD temperature of 400o C and a centerline residence time of 15 s. This experimental approach was applied in a winter field campaign and provided a direct measurement of the number concentration and size distribution for particles emitted from major pollution sources. During periods in which the contribution of biomass burning sources was dominant, more than 80% of particle number concentration remained after passing through the thermodenuder, suggesting that nearly all biomass burning particles had a nonvolatile core. These remaining particles consisted mostly of black carbon (60% mass contribution) and organic aerosol (OA; 40%). Organics that had not evaporated through the TD were mostly biomass burning OA (BBOA) and oxygenated OA (OOA) as determined from AMS source apportionment analysis. For periods during which traffic contribution was dominant 50–60% of the particles had a nonvolatile core while the rest evaporated at 400o C. The remaining particle mass consisted mostly of black carbon with an 80% contribution, while OA was responsible for another 15–20%. Organics were mostly hydrocarbon-like OA (HOA) and OOA. These results suggest that even at 400o C some fraction of the OA does not evaporate from particles emitted from common combustion processes, such as biomass burning and car engines, indicating that a fraction of this type of OA is of extremely low volatility. This work has been published as Gkatzelis, et al. [2016].
Smog chamber experiments were conducted to study the changes of the physical properties and the chemical composition of biomass burning particles as they evolve in the atmosphere. A Soot Particle Aerosol Mass Spectrometer (SP-AMS) and a Single Particle Soot Photometer (SP2) were used for the chemical characterization of the particles. An Aethalometer and a green and a blue photoacoustic extinctiometer (PAX) were used for the study of the optical properties of the particles. As smoke aged, exposed of UV light, ozone or OH radicals, organic material condensed on the preexisting particles. This coating led to an increase of the absorption of the black carbon containing particles by as much as a factor of two. The absorption enhancement of BC particles due to the coating of the BC particles with aromatic secondary organic aerosol was also studied. The coating of the biomass burning particles with anthropogenic SOA resulted in an absorption enhancement determined mainly by the changes in the particle mass concentration and not the changes of the SOA oxidation state. A core-shell Mie model could explain the corresponding absorption enhancement within experimental error in most cases [Tasoglou, et al., in press].
Conclusions:
- The assumption about mixing of the SOA components of OA appears to be robust. However, the hydrophobic POA components should probably be simulated as a separate phase.
- The additional SOA formed by the chemical aging of monoterpene and sesquiterpene ozonolysis SOA through reactions with OH results in a modest increase of the original SOA yields (less than 30% in most cases examined). This increase is higher when the SOA has been formed under high NOx conditions.
- Very oxidized SOA can be formed during the chemical aging reactions of aromatics (toluene, xylenes) with OH.
- The continued oxidation of the aromatic SOA is accompanied by a net reduction in volatility. A simple relationship has been derived linking the oxidation state of carbon and the change in effective volatility.
- The simplest parameterization of the 2D-VBS framework using the above results reproduces well the OA observations during the PEGASOS campaigns in Europe.
- We proposed standardizing a naming convention for organic aerosol classification that is relevant to laboratory studies, ambient observations, atmospheric models, and, quite importantly, the public and their leadership. This framework classifies organic material as primary or secondary pollutants and distinguishes among fundamental features important for science and policy questions including emission source, chemical phase and volatility.
- Organic aerosol exists throughout the troposphere because heterogeneous oxidation by OH radicals is more than an order of magnitude slower than comparable gas-phase oxidation.
- Condensation of anthropogenic SOA on biomass burning particles can increase their absorption up to a factor of two. A core-shell Mie model can reproduce the observed enhancement within experimental error.
- We have developed a new experimental approach that allows us to investigate chemical and physical processes perturbing ambient air in the field. The mobile experimental system used two smog chambers: one as a baseline and the second as the perturbation chamber.
- An experimental methodology was developed to measure the nonvolatile particle number concentration using a thermodenuder (TD).
- Secondary aerosol components dominate urban fine PM in most urban areas in the US and Europe. The importance of “non-traditional” sources of urban pollution (long range transport, cooking, residential and agricultural biomass burning) has been increasing while that of transportation sources is decreasing.
Journal Articles on this Report : 11 Displayed | Download in RIS Format
| Other project views: | All 12 publications | 12 publications in selected types | All 12 journal articles |
|---|
| Type | Citation | ||
|---|---|---|---|
|
|
Day MC, Zhang M, Pandis SN. Evaluation of the ability of the EC tracer method to estimate secondary organic aerosol carbon. Atmospheric Environment 2015;112:317-325. |
R835405 (2014) R835405 (Final) R835035 (Final) |
Exit Exit Exit |
|
|
Donahue NM, Chuang W, Epstein SA, Kroll JH, Worsnop DR, Robinson AL, Adams PJ, Pandis SN. Why do organic aerosols exist? Understanding aerosol lifetimes using the two-dimensional volatility basis set. Environmental Chemistry 2013;10(3):151-157. |
R835405 (2013) R835405 (2014) R835405 (Final) |
Exit Exit Exit |
|
|
Gkatzelis GI, Papanastasiou DK, Florou K, Kaltsonoudis C, Louvaris E, Pandis SN. Measurement of nonvolatile particle number size distribution. Atmospheric Measurement Techniques 2016;9(1):103-114. |
R835405 (Final) R835035 (Final) |
Exit Exit |
|
|
Hildebrandt Ruiz L, Paciga AL, Cerully KM, Nenes A, Donahue NM, Pandis SN. Formation and aging of secondary organic aerosol from toluene: changes in chemical composition, volatility, and hygroscopicity. Atmospheric Chemistry and Physics 2015;15(14):8301-8313. |
R835405 (2013) R835405 (2014) R835405 (Final) |
Exit Exit Exit |
|
|
Kostenidou E, Karnezi E, Hite Jr. JR, Bougiatioti A, Cerully K, Xu L, Ng NL, Nenes A, Pandis SN. Organic aerosol in the summertime southeastern United States: components and their link to volatility distribution, oxidation state and hygroscopicity. Atmospheric Chemistry and Physics 2018;18(8):5799-5819. |
R835405 (Final) R835403 (Final) R835410 (Final) |
Exit Exit Exit |
|
|
Murphy BN, Donahue NM, Robinson AL, Pandis SN. A naming convention for atmospheric organic aerosol. Atmospheric Chemistry and Physics 2014;14(11):5825-5839. |
R835405 (2013) R835405 (2014) R835405 (Final) |
Exit Exit Exit |
|
|
Pandis SN, Skyllakou K, Florou K, Kostenidou E, Kaltsonoudis C, Hasa E, Presto AA. Urban particulate matter pollution: a tale of five cities. Faraday Discussions 2016;189:277-290. |
R835405 (2014) R835405 (Final) |
Exit Exit |
|
|
Posner LN, Pandis SN. Sources of ultrafine particles in the Eastern United States. Atmospheric Environment 2015;111:103-112. |
R835405 (2014) R835405 (Final) R833374 (Final) R835035 (2013) R835035 (Final) |
Exit Exit Exit |
|
|
Tasoglou A, Pandis SN. Formation and chemical aging of secondary organic aerosol during the β-caryophyllene oxidation. Atmospheric Chemistry and Physics 2015;15(11):6035-6046. |
R835405 (2013) R835405 (2014) R835405 (Final) |
Exit Exit Exit |
|
|
Wang N, Kostenidou E, Donahue NM, Pandis SN. Multi-generation chemical aging of α-pinene ozonolysis products by reactions with OH. Atmospheric Chemistry & Physics 2018;18(5):3589-3601. |
R835405 (Final) |
Exit Exit |
|
|
Wang N, Jorga S, Pierce J, Donahue N, Pandis S. Particle wall-loss correction methods in smog chamber experiments. ATMOSPHERIC MEASUREMENT TECHNIQUES 2018;11(12):6577-6588. |
R835405 (Final) |
Exit Exit |
Supplemental Keywords:
Progress and Final Reports:
Original AbstractThe perspectives, information and conclusions conveyed in research project abstracts, progress reports, final reports, journal abstracts and journal publications convey the viewpoints of the principal investigator and may not represent the views and policies of ORD and EPA. Conclusions drawn by the principal investigators have not been reviewed by the Agency.