
Grantee Research Project Results
2008 Progress Report: A Novel Molecular-Based Approach for Broad Detection of Viable Pathogens in Drinking Water
EPA Grant Number: R833011Title: A Novel Molecular-Based Approach for Broad Detection of Viable Pathogens in Drinking Water
Investigators: Meschke, John Scott , Cangelosi, Gerard A.
Institution: University of Washington , Seattle Biomedical Research Institute
EPA Project Officer: Aja, Hayley
Project Period: July 3, 2006 through July 2, 2009 (Extended to August 31, 2010)
Project Period Covered by this Report: July 3, 2007 through July 2,2008
Project Amount: $597,987
RFA: Development and Evaluation of Innovative Approaches for the Quantitative Assessment of Pathogens in Drinking Water (2005) RFA Text | Recipients Lists
Research Category: Drinking Water , Water
Objective:
The objective of this research is to develop and evaluate a novel, molecular-based approach for broad detection and enumeration of viable pathogens in drinking water.Progress Summary:
Progress in this project year has been focused on WGA/WTA method development and evaluation, pre-rRNA RT-PCR, and evaluation of a flat glass device for nucleic acid extraction.
Work Progress:
Whole Genome Amplification (WGA)
In the previous project year we established the use of robust qPCR protocols for the detection and quantification of our target organisms (MAC, Aeromonas, and Adenovirus type 41). Additionally we adapted a new commercially available WGA kit for amplification of small quantities of target DNA from mock water concentrates. In the current project year, we refined the WGA method and evaluated the ability of the method to maintain relative concentrations between dilutions with and with out the presence of extraneous microbial DNA.

Figure 1. Schematic of protocol for evaluation of WGA for amplification of MAC and Aeromonas targets in water.
Mock drinking water concentrate extracts were spiked with small quantities of DNA from the target organisms: Adenovirus type 41, Aeromonas hydrophila, and Mycobacterium avium. DNA was extracted from the stocks of each target organism, and the concentration of each target determined based on qPCR, and fluorospectrophotometry using PicoGreen. Seeded extracts were amplified using the Genomeplex WGA kit (Sigma, St. Louis, MO). Pathogen-specific quantitative polymerase chain reactions (qPCRs) using TaqMan probes were used to quantify viral and bacterial targets before and after WGA. Serially diluted nucleic acid standards of known target organism concentration were included in each set of qPCR reactions to create a 4-point standard curve for quantification of sample concentrations.
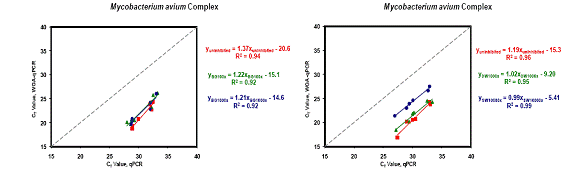
Figure 2. WGA Results for MAC. Left figure. Squares represent uninhibited samples; circles represent samples with background DNA concentrations at 100X target concentration; and triangles represent samples with background DNA at 1000X target concentrations. Right figure. squares represent uninhibited samples; circles represent samples with surface water DNA concentrations at 100X target concentration; and triangles represent samples with surface water DNA at 1000X target concentrations.
Most environmental samples contain low quantities of pathogens which may be at or below the LOD using molecular methods but, nonetheless, pose a potential health risk. One of the main goals of this project was to evaluate the application of WGA to improve detection of small quantities of pathogens. Results suggest that WGA can amplify small quantities of DNA with minimal amplification bias, while at the same time increasing the total sample volume available for subsequent analysis. Results further show that Omniplex WGA can reliably amplify target microbial DNA concentrations that are much lower than those recommended for Omniplex WGA. The Omniplex protocol recommends a minimum of 10 ng starting DNA per reaction. The input of DNA from each target organism in the current study was at most ~0.01 ng (the average target organism input in the Path A samples). However, results indicate that WGA efficiency varies with target input. According to the protocol, Omniplex WGA generates ~500 fold amplification (~2.5 log increase), but less overall amplification was seen in this study. DNA pathogens were, on average amplified by ~1-1.5 logs with WGA. Another benefit of WGA is to increase the total volume of sample available for analysis. Omniplex kits increase an initial sample of 10 ul to 75 ul of WGA product. Access to a larger amount of product makes it easier to run multiple subsequent reactions for multiple pathogen targets.
Another goal of this project was to determine if relative representation of the starting population of target DNA was maintained. Regressions were very linear, with R2 values showing good correlation considering the variability in the highly sensitive qPCR assays. Slope is indicative of the average ratio of WGA:pre-WGA target number over all of the samples.
A final goal of this project was to determine if WGA can be successfully applied to environmental samples containing mixed backgrounds of microorganisms. WGA was able to amplify target nucleic acids despite competition from background DNA. It appears that moderate concentrations of background DNA and other contaminating substances have a minor effect on amplification efficiency, while higher concentrations have more of an impact. In this study WGA successfully amplified targets of low concentration despite the presence of large of amounts of competing DNA from a natural water sample which probably contained substances which typically interfere with PCR.
Whole Transcriptome Amplification (WTA):
In the previous project year we established robust qRT-PCR protocols and completed preliminary testing of commercially available WTA, based on a combination of Multiple displacement and LMP principles, for amplification of echovirus 13. In the current year, we evaluated the lower limit of sensitivity for the WTA method relative to RT-PCR and evaluated the ability of the WTA method to maintain relative abundance of the target sequences between dilutions, with and without the presence of extraneous nucleic acids. WTA has not previously been assessed as a tool for improving molecular detection of low levels of pathogens in environmental samples.
Similar to WGA, one concern with the application of WTA to low numbers of pathogens is the requirement for a specific amount of starting RNA, as RNA quantities suggested for WTA are higher than the amount of target RNA which would be anticipated in a typical environmental sample. Using mock drinking water samples spiked with known quantities of Echovirus, this study assessed the general applicability of WTA to pathogen target amplification, the variability of WTA amplification with varying quality or quantity of target organism RNA, and the potential for inhibition of the amplification from environmental matrices and extraneous nucleic acid.
RNA was extracted using the QIAamp Viral RNA Mini Kit (QIAGEN, Valencia, CA) from echovirus 13 stocks and the quantity of nucleic acid quantified using RiboGreen on a Nanodrop 3300 fluorospectrometer. Various concentrations of viral RNA were spiked into mock water concentrate extracts and WTA applied. WTA was conducted using the TransPlex Whole Transcriptome Amplification Kit (Sigma-Aldrich, St. Louis, MO). Pre-WTA and post WTA samples were quantified by qRT-PCR using a FAM-labeled TaqmanÒ probes to quantify the impact of WTA on target nucleic acid concentration and examine the relative abundance of target between dilutions. Serially diluted nucleic acid standards of known target organism concentration were included in each set of qPCR reactions to create a 4-point standard curve for quantification of sample concentrations.
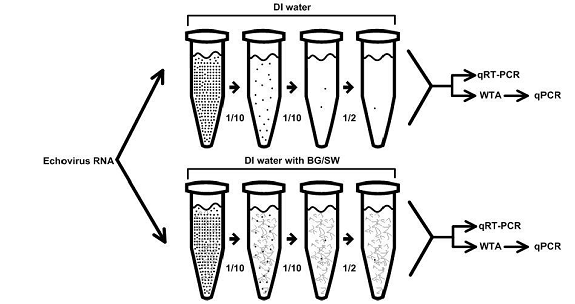
Figure 3. Schematic of protocol for WTA evaluation.
WTA sample amplification ranged from ~2-4 log increase in target copy number. Additionally there was an increase in the percentage of positive low EV titer samples after WTA. Transplex WTA successfully amplified viral RNA in quantities smaller than intended for the kit. Transplex WTA recommends input of 50 ng RNA, but notes that <5 ng is usable. Total EV RNA concentrations were <<5 ng, based on qRT-PCR results. Another benefit of WTA is to increase the total volume of sample available for subsequent analysis (e.g. qPCR, microarray, etc.). Transplex kits increase an initial sample of 10 ul to 375 ul of WTA product. Access to a larger amount of product makes it easier to run assays for multiple pathogen targets.
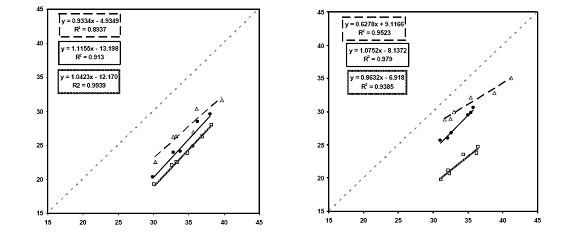
Figure 4. WTA Results. Left figure. squares represent uninhibited samples; circles represent samples with background DNA concentrations at 100X target concentration; and triangles represent samples with background DNA at 1000X target concentrations. Right figure. squares represent uninhibited samples; circles represent samples with surface water DNA concentrations at 100X target concentration; and triangles represent samples with surface water DNA at 1000X target concentrations.
Slope of the regression lines were indicative of the average ratio of WTA:pre-WTA target number. Variation in the slope (variation in amplification efficiency) was seen depending on the amounts and types of background added to the experiments. WTA was able to amplify target nucleic acids despite competition from background DNA. It appears that moderate concentrations of background DNA have a minor effect on amplification efficiency, while higher concentrations have more of an impact. Though inhibition was difficult to quantify, WTA successfully amplified targets of low concentration despite the presence of large of amounts of competing DNA from a natural water sample which contained substances which typically interfere with PCR.
In conclusion, results from these experiments demonstrate that WTA can be effectively applied to mixed microbial communities, such as those that might be found in drinking water.
pre-rRNA RT-PCR:
As a molecular method for detecting microorganisms in samples, the polymerase chain reaction (PCR) is fast and sensitive. However, its utility is limited in part by its inability to distinguish viable pathogen cells in samples from inactivated cells and free nucleic acid fragments. To address this limitation we developed PCR-based tests for bacterial ribosomal RNA precursor (pre-rRNA) molecules.
Pre-rRNAs are intermediates in rRNA synthesis generated by rapid nucleolytic cleavage of polycistronic rRNA operon transcripts. Leader and tail fragments are removed in slower reactions tied to ribosome assembly, yielding the mature rRNA subunits. Ribosome biosynthesis is energy-expensive and therefore tightly regulated according to growth needs. In growing bacterial cells, pre-rRNAs account for a large fraction of total rRNA molecules. They are more abundant and easier to detect than even the most strongly-expressed mRNA molecules. Upon cessation of growth, pre-rRNA synthesis ceases but its processing continues, resulting in active and substantial drainage of pre-rRNA pools. The present study exploited a more consistent factor in pre-rRNA pool maintenance, namely the rapid replenishment of pre-rRNA in growth-limited cells that sense new nutrients in their environments. Bacterial cells were distinguished from inactivated cells by using RT-qPCR to measure species-specific pre-rRNA in water samples that have been briefly stimulated with nutrients relative to control samples that were not stimulated with nutrients. This ratiometric approach, was developed for two water-associated bacterial pathogens, the rapidly growing gram negative species Aeromonas hydrophila and the slowly growing actinomycete Mycobacterium avium.

Figure 5. Schematic of pre-rRNA processing. Stars indicate region targeted for primer design.
Specific primer sets targeting the 5’ pre-rRNA leader region of the pre-rRNA were developed for both A. hydrophila and M. avium, based on the assumption that this promoter-proximal region would have the greatest abundance in cells that are actively transcribing rRNA. Primer sets were designed to straddle the 5’ mature rRNA terminus. Primers for cDNA synthesis and reverse PCR primers recognized semi-conserved sequences within the mature rRNA. Forward primers recognized species-specific sequences within the 5’ leader region. Therefore, successful amplification required intact pre-rRNA molecules as templates for cDNA generation.
PCR reactions with gel electrophoresis readouts consistently yielded products of the expected sizes when applied to nucleic acid from 15 isolates of M. avium subspecies homissuis and 4 isolates of M. intracellulare. These two closely-related species are the most significant human pathogens within the complex. No products were observed when the reactions were applied to nucleic acid from other Mycobacterium species, namely M. terrae, M. gastri, M. smegmatis, M. nonchromogenicum, M. phlei, and M. vaccae.
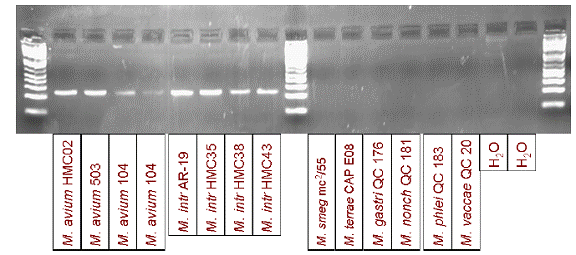
Figure 6. Results of MAC primer evaluation.
A. hydrophila strain ATCC 7966 cells and M. avium subspecies hominissuis strain HMC02 were grown to early stationary-phase in Nutrient Broth and Middlebrook 7H9 broth supplemented with 10% ADC, respectively. Cells were pelleted, washed with autoclaved tap water (ATW), and resuspended in ATW. To drain pre-rRNA pools under conditions that simulate water supplies, these suspensions in ATW were incubated for 7-14 days. Paired portions each suspension were then diluted 100-fold into Nutrient Broth (stimulated aliquot) or ATW (control aliquot). These suspensions were incubated for 60 minutes to enable pre-rRNA stimulation, a portion of each of these suspensions was pelleted by centrifugation, RNA extracted and subjected to qRT-PCR using specific primers. A postive pre-rRNA stimulation ratio was indicative of the presence of viable cells. Time course experiments (N > 5 for each organism) showed that pre-rRNA stimulation was very rapid in viable cells of both A. hydrophila and M. avium. Under the conditions of our experiments, 15 minutes of stimulation was adequate for consistent measurement of pre-rRNA upshift in A. hydrophila (Figure 1A). Approximately 4 hours was required for maximal pre-rRNA stimulation in M. avium, a slow-growing organism with a generation time of >20 hours (Figure 1B). For both organisms these time periods are <1 generation time.
Additional experiments were performed to assess the specificity of ratiometric pre-rRNA analysis (RPA) for viable cells, sodium hypochlorite was used to generate A. hydrophila cell suspensions with varying ratios of viable and inactivated cells. A. hydrophila cells, diluted in ATW, were exposed to varying concentrations to yield differentially chlorinated cell suspensions with varying ratios of active and inactivated cells. These cells were then assayed for pre-rRNA stimulus as described above and a second portion of the nutritionally stimulated cells was similarly pelleted and plated to enumerate colony forming units (cfu). Additionally, in some experiments, genomic DNA in stimulated and control samples was also quantified by qPCR. This allowed us to assess the specificity of RPA to viable cells, in comparison to the specificity seen with qPCR of DNA.
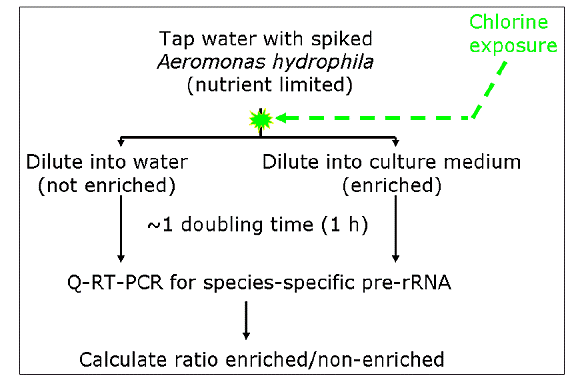
Figure 7. Schematic of protocol for evaluation of RPA approach to determine viability.
Samples with cultivatable bacteria typically demonstrated pre-rRNA stimulation ratios of >2 ± 1 SD, while samples with no detectable viable cells (0% viability) typically exhibited pre-rRNA stimulation ratios that were not statistically greater than 1.0. There was no difference between DNA signals in nutritionally stimulated and control aliquots.

Figure 8. Aeromonas PSR, genomic DNA, and culture after chlorination
As a common inhabitant of surface waters, A. hydrophila was a convenient model for field testing RPA. Water samples were collected from fresh water sites (Lake Washington and Lake Union) and a salt water site (Puget Sound) in Seattle, WA. A portion of each sample was autoclaved to generate an inactivated control. Autoclaved and non-autoclaved samples (300 mL each) were concentrated by filtration and assayed by culture and RPA. The fresh water samples yielded viable counts of A. hydrophila ranging from 280 to 798 cfu/mL. All of them exhibited positive RPA signals (Table 3). All autoclaved samples yielded no cfu and no A. hydrophila pre-rRNA was detected in these samples. The salt water sample had a viable count of 6 cfu/mL A. hydrophila, however no A. hydrophila pre-rRNA was detected in either stimulated or non-stimulated samples, with or without autoclaving. Therefore, in its current form RPA applied to natural samples had a detection limit that fell between 6 and 280 cfu/mL.
The results of this study support the feasibility of using RPA to improve the specificity of nucleic acid based detection of viable pathogens in environmental samples. The method is robust and built upon a physiological trait of all known bacteria. It may prove broadly useful in food and water safety analysis, either by itself or as an adjunct to other tools such as PMA and EMA qPCR methods. Further studies are needed to evaluate the performance of RPA in the contexts of diverse bacterial species, sample types, and disinfection methods.
Nucleic acid Extraction Cards:
In this project year we began collaborating with a company, Blood Cell Storage Incorporated (BCSI, Seattle, WA), who have developed a flat glass based fluidic device for nucleic acid extraction and purification. A sample is mixed with an extraction buffer and then circulated through a serpentine fluidic channel on the flat glass device. The extracted nucleic acid binds firmly to the flat glass surface and can then be washed and eluted into a smaller volume. This system was initially designed for extraction of nucleic acid from blood and plasma samples for contamination testing and has potential benefits over existing nucleic acid extraction formats for water samples in that it may be easily automated, be adapted to a larger initial sample volume, easily wash away contaminating substances.

Figure 9. Photograph of BCSI flat glass fluidic device for nucleic acid extraction.
In this project year, initial experiments were performed to examine the efficiency and dynamic range of the BCSI device for bacterial DNA. A model assay for detecting E. coli DNA in water, based on a published real-time PCR assay for the detection of E. coli DNA(Frahm and Obst, Journal of Microbiological Methods 52:125, 2003), was adopted for the experiments. The assays utilized TAQMAN probes (Applied Biosystems, Foster City, CA) for detection of amplification and compatible fluorescent DNA intercalating dyes as a means of tracking carrier DNA. This assay was modified to test the lower limit of detection when the prototype flat glass device was used to re-purify bacterial DNA, and the use of carrier DNA was evaluated.
E. coli bacterial DNA from strain ATCC11303 type B ( MP Biomedicals, Solon, OH), with original concentration of 1mg/mL in water, was used in the experiments. Ten-fold serial dilutions were prepared ranging from 1 mg/mL to 1 pg/mL in DEPC-treated water (FLUKA: Sigma-Aldrich, St. Louis, MO). The E. coli DNA standard curve generated showed the lower limit of detection of the assay to be as low as18 pg/mL. DNA isolated from the device after washing was very similar to that of the DNA before purification device, suggesting the efficiency of the device for binding and elution of bacterial DNA was quite good.
The use of carrier nucleic acid in detection of low levels of DNA was examined in the real-time PCR reaction. The use of the carrier DNA was expected to normalize fluorescence signals during probe-based detection reactions where various quantities of DNA are amplified and to allow for direct, in-process quantification of the extracted nucleic acids on the device (using compatible DNA intercalating dyes). Different carrier molecules were tested at set concentrations by titrating in carrier DNA along with the template, and a standard curve (dilution series) of E. coli DNA was run with the probe for each of the individual concentrations. Fish DNA, human DNA, yeast tRNA (obtained from Sigma-Aldrich), and synthetic dA/dT (obtained from Sigma-Aldrich) were evaluated at final concentrations ranging from 1 ng/mL to 1 mg/mL. The appropriate dA/dT concentration to be used in this assay was determined to be 30 ng/ml, and for tRNA, 10 ng/ml. Genomic DNA was found to interfere with the PCR assays, probably due to non-specific binding of the primers resulting in greatly reduced sensitivity to E. coli DNA.
The experiments described point to the feasibility of using the BCSI device for efficient extraction and purification of bacterial nucleic acid and the use of a synthetic carrier DNA molecule to track the DNA extraction process using a fluorescent intercalating dye followed by direct downstream processing by real-time PCR for pathogen detection in the extracted water samples. Since the pathogen DNA would only be present in trace amounts, the carrier DNA helps validate the successful extraction of nucleic acids on the BCSI device.
Future Activities:
In the third year of the project we plan to:
o Conduct filter performance and recovery experiments
o Continue nucleic acid extraction work on the BCSI microfluidic cards
o Begin development of ICC-qRT-PCR methods for detection of viable +ssRNA viruses.
o Submission of a manuscript on the pre-rRNA method.
Journal Articles:
No journal articles submitted with this report: View all 16 publications for this projectSupplemental Keywords:
Whole Genome Amplification, pre-rRNA, Aeromonas, MAC, Adenovirus, Echovirus, nucleic acid extraction, filteration., RFA, Scientific Discipline, Water, Environmental Chemistry, Drinking Water, Environmental Engineering, Environmental Monitoring, analytical methods, monitoring, pathogens, polymerase chain reaction, drinking water contaminants, drinking water monitoringProgress and Final Reports:
Original AbstractThe perspectives, information and conclusions conveyed in research project abstracts, progress reports, final reports, journal abstracts and journal publications convey the viewpoints of the principal investigator and may not represent the views and policies of ORD and EPA. Conclusions drawn by the principal investigators have not been reviewed by the Agency.