
Grantee Research Project Results
2008 Progress Report: Sensitivity of Heterogeneous Atmospheric Mercury Processes to Climate Change
EPA Grant Number: R833375Title: Sensitivity of Heterogeneous Atmospheric Mercury Processes to Climate Change
Investigators: Schauer, James J. , Griffin, Robert J. , Shafer, Martin M. , Holloway, Tracey
Institution: University of Wisconsin - Madison , University of New Hampshire
EPA Project Officer: Chung, Serena
Project Period: February 15, 2007 through February 14, 2010 (Extended to February 14, 2011)
Project Period Covered by this Report: February 15, 2008 through February 14,2009
Project Amount: $899,731
RFA: Consequences of Global Change For Air Quality (2006) RFA Text | Recipients Lists
Research Category: Air , Climate Change
Objective:
The overall goal of the proposed project is to quantify the impact of climate change on key atmospheric processes that control the fate of mercury during transport from emission to deposition. Efforts are being directed at building on the existing scientific understanding of atmospheric mercury processes by examining the incremental impact of climate change variables on heterogeneous atmospheric mercury oxidation and depositional processes.
The goal is being realized by achieving the following objectives:
Progress Summary:
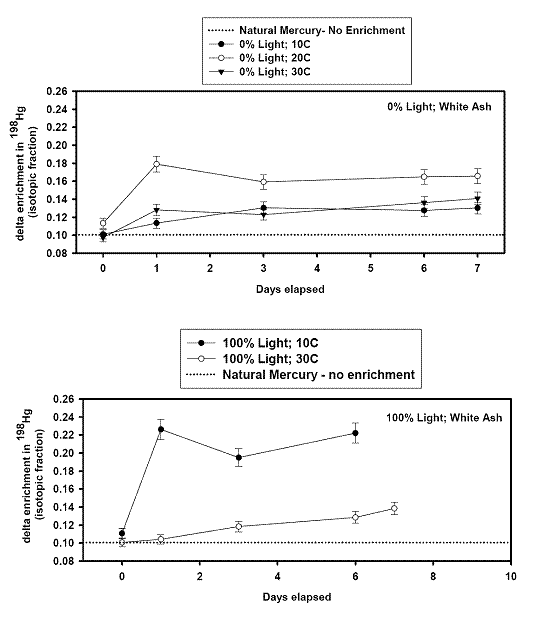
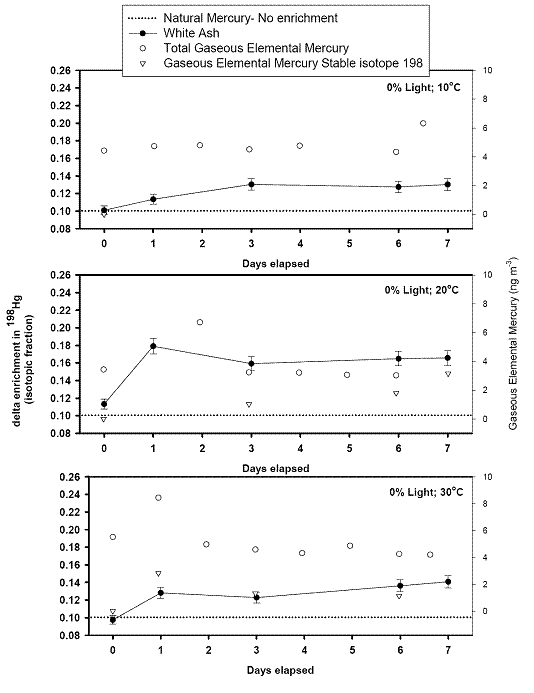
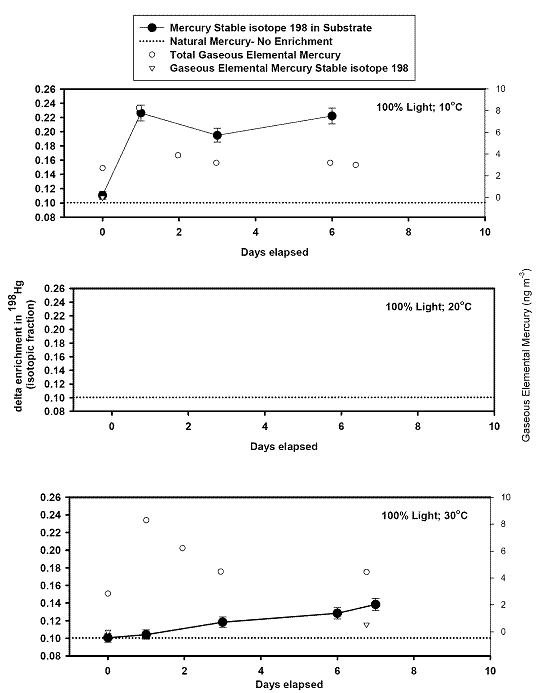
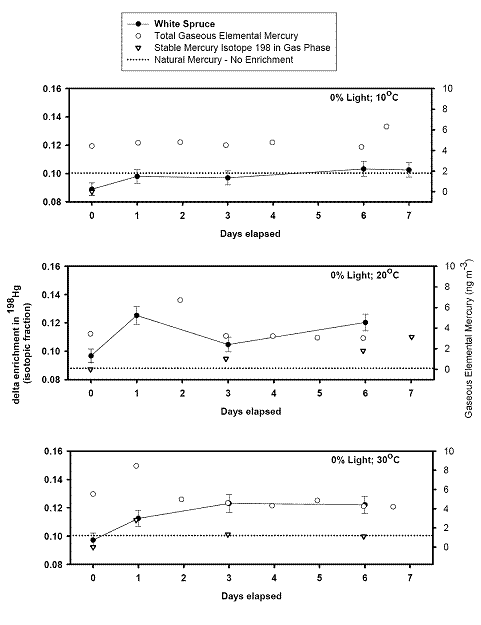
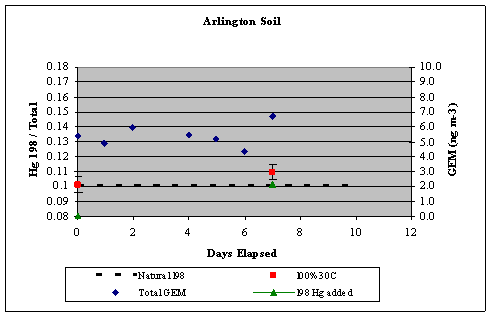
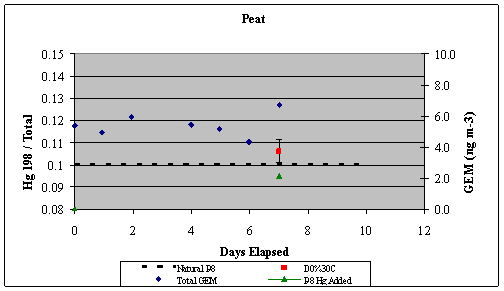
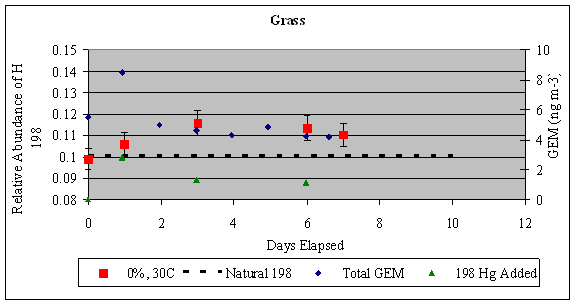
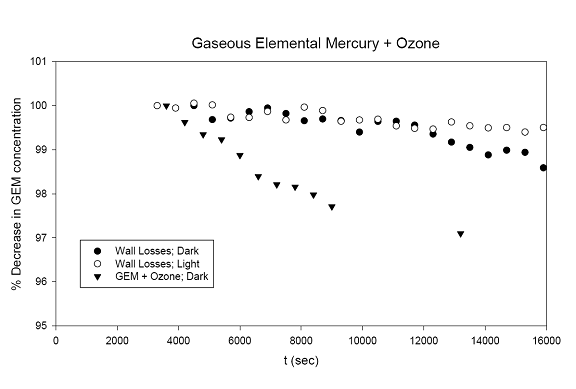
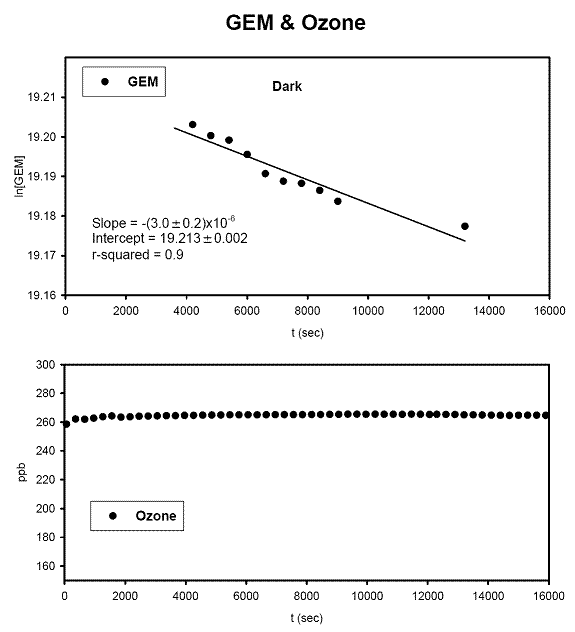
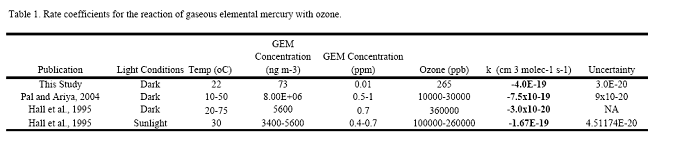
left align">Figure 9. Analysis of the GEM + Ozone reaction system results for a pseudo first order rate coefficient that could then be turned into a second order rate coefficient and compared with literature values (Table 1).
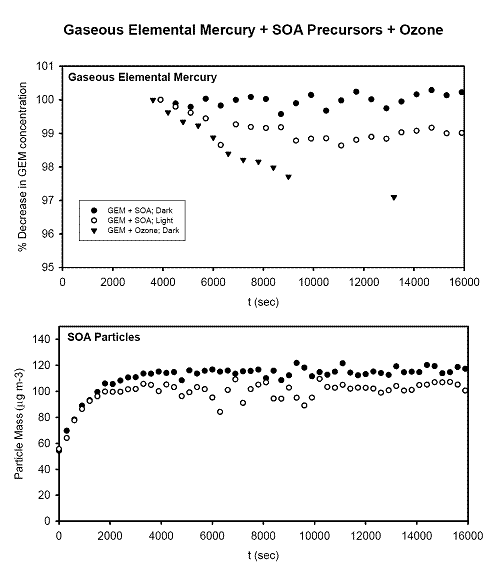

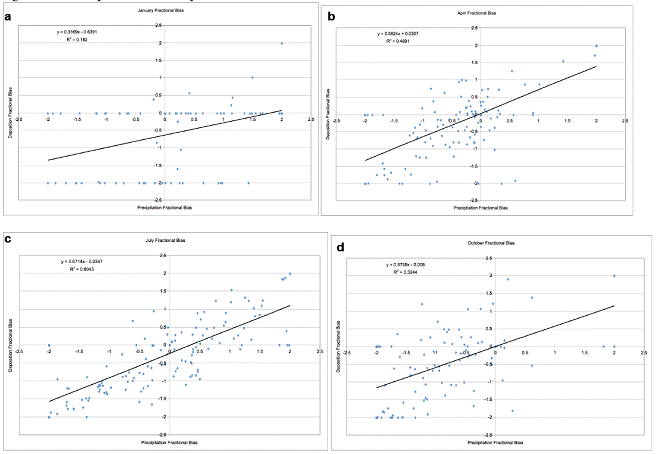
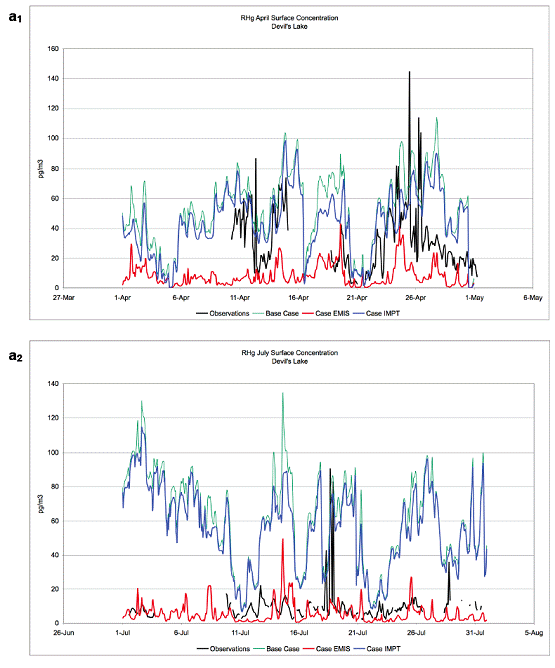
Future Activities:
We will carefully consider our results to date, and design a defensible plan to evaluate the response of model processes to changes in climate variables including temperature, humidity, cloud cover, and precipitation. We are aware that, if model errors dominate concentration and deposition signals, care must be taken in assessing climate sensitivity.
References:
Journal Articles:
No journal articles submitted with this report: View all 4 publications for this projectSupplemental Keywords:
RFA, Air, climate change, mercury depositionProgress and Final Reports:
Original AbstractThe perspectives, information and conclusions conveyed in research project abstracts, progress reports, final reports, journal abstracts and journal publications convey the viewpoints of the principal investigator and may not represent the views and policies of ORD and EPA. Conclusions drawn by the principal investigators have not been reviewed by the Agency.